Det craniodiaphyseal dysplasi er en medfødt knoglesygdom, der er forbundet med hyperostose og sklerose i ansigtsskallen. Årsagen er en genetisk mutation i de gener, der hæmmer knoglestrukturen. Terapi er symptomatisk og fokuserer på at stoppe sygdommen i at udvikle sig.
Hvad er craniodiaphyseal dysplasi?

© crevis - stock.adobe.com
Ved hyperostoser øges knoglestoffet på en patologisk måde. Hovedostosen af kraniet er en gruppe af sygdomme, der er relateret til en sådan stigning i knoglestoff i kraniet. Som craniodiaphyseal dysplasi er kendetegnet ved en medfødt hyperostose af kraniet og er en knoglesygdom.
Den australske læge John Halliday beskrev først sygdommen i midten af det 20. århundrede. Hyppigheden er angivet med en forekomst på mindre end et tilfælde hos 1.000.000 mennesker. Dette gør knoglesygdomme til en ekstremt sjælden dysplasi af kraniet.
Komplekset med hyperostose og stenose i ansigts- og kraniale knogler er nu blevet sporet tilbage til en genetisk årsag. På grund af de få dokumenterede tilfælde hidtil er ikke alle forbindelser mellem sygdommen blevet afklaret. Af denne grund er terapimulighederne også begrænset i øjeblikket.
årsager
I et stort antal tilfælde forekommer craniodiaphyseal dysplasi ikke sporadisk, men med familiær akkumulering. Både den autosomale recessive og den autosomale dominerende arvsmåde er blevet identificeret som tilstanden for arv for sygdommen. Den autosomale dominerende form af sygdommen er baseret på en ny mutation i SOST-genet. Genet er lokaliseret på lokalitet 17q21.31 og betragtes som en af de vigtigste hæmmere af knogledannelse.
Mutationen af SOST-generne er ansvarlig for et stort antal af arvelige knoglesygdomme, såsom VDB. I tilfælde af en mutation kan genet ikke længere udføre sine inhiberende funktioner, og knoglestrukturen sprøjter. Dette adskiller grundlæggende hyperostose af craniodiaphyseal dysplasi fra andre hyperostoser.
De fleste af disse sygdomme er baseret på en dysfunktion af osteoklasterne eller osteoblasterne. Den genetiske disposition anses for at være bevist i forbindelse med sygdommen. Hvilke andre faktorer, der spiller en rolle i sygdommens begyndelse, er ikke blevet afklaret endeligt.
Symptomer, lidelser og tegn
Det kliniske billede af craniodiaphyseal dysplasi er kendetegnet ved forskellige kliniske kriterier, der allerede er manifesteret i spædbarnet. Berørte spædbørn har normalt stærkt forhindrede næseåbninger, hvilket kan forårsage problemer med at trække vejret. I det senere sygdomsforløb er der i de fleste tilfælde fuldstændig hindring af næsevejene.
Ofte blokeres patientens tårekanaler efter dette fænomen. På underkæben af de fleste af de ramte, dannes gradvis voksende næsebulder af bony substans. Hyperostosen i ansigtsskallen skrider frem og udvikler sig til leontiasis ossea. I de fleste tilfælde forstyrres eller udsættes patientens tandudvikling. Det indre af kraniet indsnævres, efterhånden som sygdommen skrider frem.
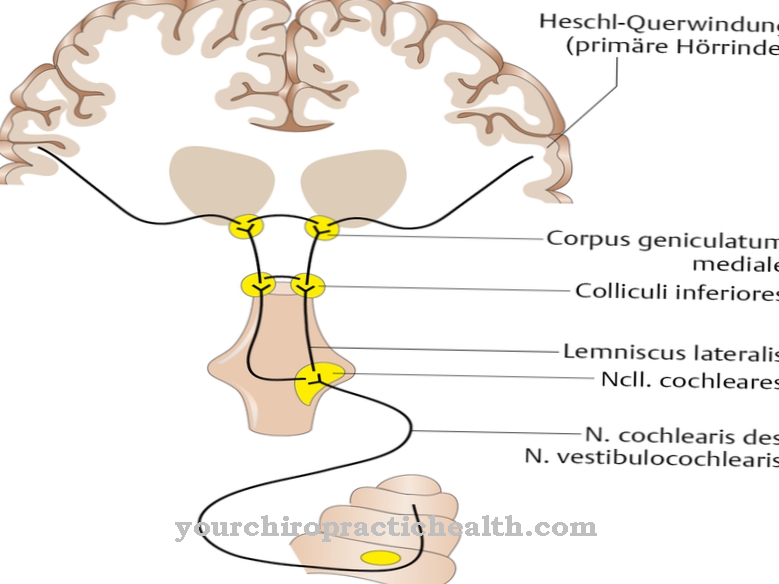
Begrænsningerne påvirker også foraminaen og forårsager fortløbende optisk atrofi. Dette kan ledsages af symptomer som høretab og mere eller mindre alvorlig hovedpine. I nogle tilfælde, da det indre af kraniet bliver mere smalt, lider patienterne også af anfald. Akslerne i de lange rørformede knogler udvides i stigende grad.
Diagnose & sygdomsforløb
Den tidligst mulige diagnose og efterfølgende behandling forbedrer prognosen for patienter med craniodiaphyseal dysplasi betydeligt. Lægen har formodentlig mistanke om hyperostose fra en visuel diagnose. Billedbehandlingsprocedurer betragtes som det vigtigste diagnostiske værktøj. For eksempel viser en røntgenstråle ekstrem hyperostose og sklerose af alle kranier.
Kravebenene eller ribbenene kan forekomme dilaterede i billeddannelsen. De manglende membraner på de lange knogler skiller sig tydeligt ud. En afgrænset, ikke-fortykket cortex passer også ind i det kliniske billede. Med hensyn til differentiel diagnose skal der skelnes fra sygdomme som Engelmanns syndrom. Molekylærgenetiske analyser er især velegnede til en sådan differentieret diagnose. Engelmann-syndrom viser ændringer i TGFB1-genet ved mutationsanalyse, mens craniodiaphyseal dysplasi påvirker SOST-genet.
Komplikationer
Craniodiaphyseal dysplasi er en sjælden, genetisk bestemt knoglesygdom. Symptomet manifesterer sig direkte på ansigtsskallen gennem en kraftig stigning i knoglestof med ledsagende sklerose. Den genetiske mutation er allerede tydelig i spædbarnet baseret på formen af kraniet og forkert lagt næsepassager, hvilket kan forårsage truende vejrtrækningsproblemer.
De resulterende konsekvenser af craniodiaphyseal dysplasi bringer den berørte patient adskillige livsbegrænsende komplikationer fra spædbarnet. Hvis der ikke er rettidig klinisk intervention, vil den overskydende knoglevækst udvikle sig. Det indre af kraniet indsnævres, og rækkerne med tænder dannes ikke tilstrækkeligt. Det fortykkende knoglemateriale indsnævrer øregangen, og der er risiko for hørselsnedsættelse og endda høretab.
Der er en stigende mangel på plads i kranialhulen, og knogleaflejringer trænger ind i hjernen. Alvorlig hovedpine, anfald, ansigtslammelse og epilepsi opstår samt en reduktion eller regression af de mentalt erhvervede færdigheder. Forældre, hvis børn er påvirket af craniodiaphyseal dysplasi, bør derfor søge kliniske forholdsregler på et tidligt tidspunkt.
Efter billeddannelsesundersøgelsen finder den differentielle diagnose sted inden for omfanget af de givne muligheder. Der er i øjeblikket ingen baseterapi for craniodiaphyseal dysplasi. Der gøres forsøg på at begrænse den ukontrollerede udvikling af knoglevækst og dens konsekvenser. Forskellige medikamenter såvel som en kalkreduceret diæt fra spædbarnet og fremover vil hjælpe den syge med at reducere symptomerne.
Hvornår skal du gå til lægen?
Craniodiaphyseal dysplasi diagnosticeres ofte umiddelbart efter fødslen. Hvis dette er tilfældet, vil den ansvarlige læge straks informere forældrene og derefter starte behandling direkte. Hvis dysplasien er mindre udtalt, stilles diagnosen af forældrene. Et lægebesøg angives, hvis den nyfødte har åndedrætsbesvær eller vandige øjne. Eksterne abnormiteter såsom de typiske misdannelser i ansigt og tænder indikerer også en sygdom, der skal afklares og behandles.
Forældre, der oplever tegn på høretab eller anfald hos deres barn, skal kontakte en læge. Det samme gælder, hvis barnet ofte klager over hovedpine eller ser ud til at være stærk smerte. Under behandlingen skal barnet regelmæssigt præsenteres for en læge. Dette vil sikre, at opsvinget går uden komplikationer. Da craniodiaphyseal dysplasi er forbundet med en række symptomer, kan behandling tage måneder eller endda år. Den praktiserende læge vil konsultere andre specialister til dette formål, altid afhængigt af symptomer og klager. Typisk er neurologer, internister, ørespecialister, kirurger, fysioterapeuter og psykologer involveret i behandlingen.
Behandling og terapi
En kausal terapi for patienter med craniodiaphyseal dysplasi findes endnu ikke. En sådan terapi kan være tænkelig i fremtiden gennem genterapimetoder. I øjeblikket kan sygdommen imidlertid kun behandles symptomatisk. Hovedmålet med alle terapeutiske foranstaltninger er at stoppe den overdreven knoglevækst. Der er forskellige skridt at tage.
Forløbet af sygdommen kan for eksempel stoppes med medicin. Calcitriol og calcitonin bruges mest som medicin. Da knoglestrukturen er afhængig af calcium, kan en kalkreduceret diæt også give mening. Denne særlige diæt skal bruges på lang sigt og ideelt ledsage patientens hele liv.
Lægemiddelbehandlingen af patienter med den kunstige glukokortikoidprednison har også vist positive effekter. Jo tidligere behandling er startet, jo mere lovende er udsigterne. Med ekstrem tidlig behandling kan hyperostose bringes til stilstand i de første leveår. På denne måde reduceres de efterfølgende symptomer drastisk.
Under visse omstændigheder kan der også foretages kirurgiske korrektioner som en del af terapien. Imidlertid giver sådanne rettelser normalt ingen mening, før sygdomsforløbet er bragt under kontrol.
Outlook og prognose
Ved medfødt, men meget sjælden craniodiaphyseal dysplasi, er der en uoprettelig genetisk mutation. Derfor er prognosen for de berørte ikke særlig god. De medicinske fagfolk kan kun forsøge at behandle symptomerne og følgerne af den øgede knoglevækst i hovedområdet. Terapi kan kun forsinke sygdomsforløbet. Ved craniodiaphyseal dysplasi er stigningen i knoglesubstans ustoppelig.
Da nutidens terapimuligheder ikke kan vende den underliggende mutation i embryotrinnet, vil andre generationer af de berørte lide af det. En familiær akkumulering er mærkbar ved craniodiaphyseal dysplasi. Symptomerne forbundet med craniodiaphyseal dysplasi kan allerede ses hos barnet. Da alle knogleadhæsioner finder sted i kraniet, påvirkes den øvre luftvej samt hørelse eller syn af dem.
Derudover påvirkes det indre af kraniet i stigende grad af knogledannelse. Dette begrænser de terapeutiske tilgange til de efterfølgende klager. Jo tidligere diagnosen kan stilles, jo bedre er den langsigtede prognose. En diæt med lavt calciumindhold forhindrer stigende knoglevækst. Derudover kan passende medicinering og prednison administreres allerede i spædbarnet.
En tværfaglig behandlingsstrategi opnår de bedste resultater. Kirurgisk indgreb i craniodiaphyseal dysplasi giver kun mening, hvis udviklingen af sygdommen med succes er indeholdt.
forebyggelse
Indtil videre er der ingen forebyggende foranstaltninger mod craniodiaphyseal dysplasi. Sygdommen er en genetisk sygdom, der er forbundet med en familiær disposition. Derfor kan kun molekylær genetisk rådgivning bruges som en slags forebyggende foranstaltning.
Efterbehandling
I de fleste tilfælde har den påvirkede meget få opfølgende foranstaltninger til rådighed. I nogle tilfælde kan dette endda være fuldstændigt begrænset, så den berørte er afhængig af rent symptomatisk behandling af sygdommen. Selvhelende kan ikke forekomme, fordi det er en genetisk sygdom.
Derfor, hvis den pågældende ønsker at få et barn, bør de foretages en genetisk undersøgelse og rådgivning, så sygdommen ikke gentager sig hos børnene. Selve behandlingen udføres normalt ved hjælp af forskellige medikamenter, der permanent kan lindre og begrænse symptomerne. Det er altid vigtigt at sikre, at det tages regelmæssigt, hvorved den korrekte dosering også skal overholdes.
For børn bør forældrene især kontrollere, at de bliver taget og brugt korrekt. Regelmæssig kontrol af en læge er også nødvendig for at kunne kontrollere sygdommens tilstand permanent. De fleste misdannelser kan korrigeres ved kirurgiske indgreb. Mange af de berørte er også afhængige af psykologisk støtte fra deres egen familie i deres hverdag, hvilket har en positiv effekt på det videre sygdomsforløb. Som regel reducerer denne sygdom ikke patientens forventede levetid.
Du kan gøre det selv
I tilfælde af craniodiaphyseal dysplasi har den berørte patient kun begrænsede effektive foranstaltninger til rådighed, der har en positiv effekt på sygdomsforløbet. Først og fremmest er en passende behandling af craniodiaphyseal dysplasi af et team af specialister. Sygdommen begynder at manifestere sig i spædbarnet, så det primært er forældrene, der bidrager til livskvaliteten for de berørte børn. I tilfælde af ophold på barnets patienter er det fornuftigt, hvis forældrene er til stede på hospitalet, og barnet får følelsesmæssig støtte som et resultat.
I løbet af sygdommen er der ofte forstyrrelser i udviklingen af tænderne, så patienterne ofte er afhængige af ortodontisk terapi. Dit eget samarbejde kræves også, når det kommer til at bære seler. Der er også bevis for, at en diæt med lavt calciumindhold kan bremse progressionen af craniodiaphyseal dysplasi. Også her har patienterne stort spillerum med hensyn til deres samarbejde og dermed deres livskvalitet.
På grund af vejrtrækningsproblemerne forlader patienterne visse sportsgrene, men praktiserer også styrkende øvelser derhjemme hos en fysioterapeut, hvis dette er medicinsk tilladt. Børn med craniodiaphyseal dysplasi får tilstrækkelig uddannelse i specialskoler.






.jpg)



.jpg)