Det Jeg cellesygdom er en lyosomal mucolipidose. Årsagen til opbevaringssygdommen er en mutation af GNPTA-genet med genet locus q23.3 på kromosom 12. Symptomatisk behandling udføres hovedsageligt ved indgivelse af bisphosphonater.
Hvad er jeg cellesygdom?

© red150770 - stock.adobe.com
Opbevaringssygdomme er kendetegnet ved afsætning af forskellige stoffer i celler og organer i den menneskelige krop. Det er en heterogen gruppe af sygdomme, der kan opdeles i flere underformer. Foruden glycogenoser, mucopolysaccharidoser og lipidoser, adskiller medicinen sig afhængigt af det deponerede stof, sfingolipidoser, hæmosideroser og amyloidoser.
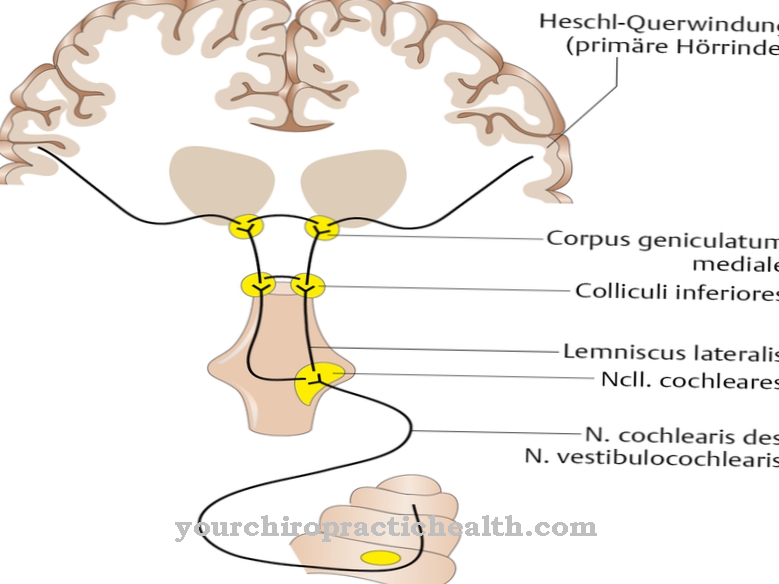
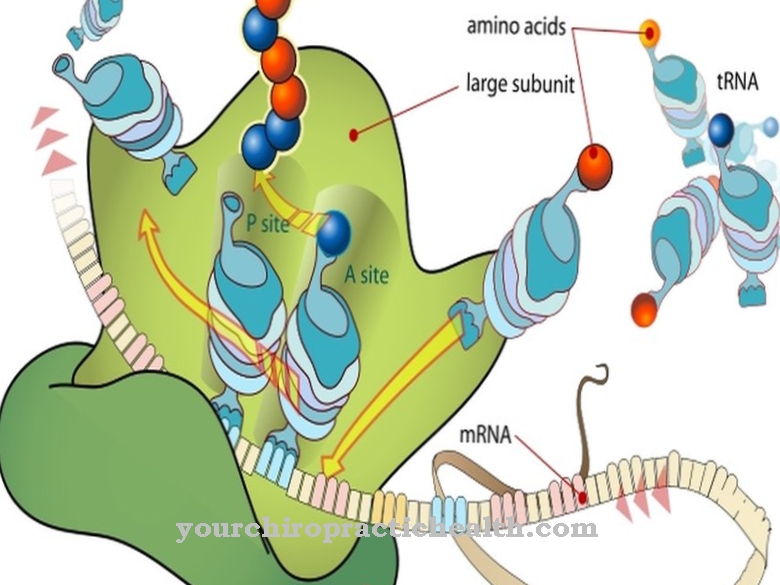
Lysosomale opbevaringssygdomme påvirker lysosomerne. Disse er små membranovertrukne celleorganeller i eukaryoterne. Lysosomer dannes af Golgi-apparatet og er udstyret med hydrolytiske enzymer og phosphataser. Ved hjælp af deres enzymer bør de primært fordøje fremmedstoffer og kroppens egne stoffer.
I-celle sygdom er en lyosomal mucolipidose med to forskellige undertyper. Leroy og DeMars dokumenterede først sygdommen i 1960'erne og pegede på dens lighed med mucopolysaccharidosis type I, kendt som Hurlers sygdom. Sygdommens navn kommer fra fibroblastindeslutningerne, de såkaldte inklusionsceller, i patientens hud.
årsager
Årsagen til I-celle sygdommen ligger i en mangel på aktivitet af N-acetylglucosaminyl-1-phosphotransferase. Dette enzyms begrænsede aktivitet forhindrer, at en stor del af de lysosomale enzymer trænger ind i lysosomets indre. Reguleringen af de lysosomale enzymer er formet af aktiviteten af phosphotransferase.
Det muliggør syntese af et sorteringssignal i en sund organisme. Denne proces forstyrres ved I-cellesygdom. Der er derfor ingen mærkning med mannose-6-phosphat. Af denne grund sorteres de lysosomale enzymer ikke længere korrekt og migrerer ind i den ekstracellulære matrix på en ukontrolleret måde via plasmamembranen.
Årsagen hertil er en mutation i GNPTAB-genet. Det fjerner funktionaliteten af N-acetylglucosaminyl-1-phosphotransferase og dermed evnen til at katalysere syntesen af mannose-6-phosphat. Transporten af lysomale enzymer er så forstyrret. N-acetyl-glucosamin-1-phosphotransferase består af underenhederne alfa, beta og gamma. De er kodet på to gener.
Den arvelige I-celle sygdom påvirker GNPTA-genet på kromosom 12. Der er en mutation i q23.3-genlokuset. For den sjældne sygdom gives en forekomst på ca. 0,3: 100.000. Arv er underlagt autosomal recessiv arv. Begge forældre skal derfor bære det defekte gen for at overføre sygdommen.
Symptomer, lidelser og tegn
I de fleste tilfælde kan symptomerne på I-celle sygdomme observeres umiddelbart efter fødslen eller et par måneder senere, og deres egenskaber ligner dem ved Hurlers syndrom. I modsætning til patienter med Hurler-syndrom, udviser patienter med I-cellesygdom ingen mukopolysaccharidudskillelse.
De individuelle symptomer på sygdommen udsættes for et stort antal variationer. Kornfeld og Sly opsummerer kliniske træk ved skelettet, indre organer, øjne, hud, centralnervesystem og ansigt. Skelettet er så ofte påvirket af kyphoscoliosis og hoftedislokationer.
Klubfødder, ledkontraktioner og deformiteter i ryghvirvlerne kan også være til stede. Det samme gælder for kortstatus og dysostosemultiplex. Sygdommen kan manifestere sig i de indre organer i form af hepatosplenomegaly og cardiomegaly eller hjertesygdom. Patientens ansigt har grove træk.
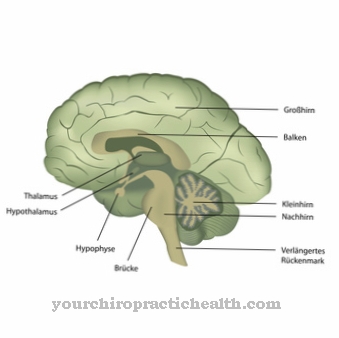
Exophthalmos, hyperplastisk tandkød eller scaphocephaly er typiske symptomer. Også karakteristisk er en åben mund og en dybt nedsænket næse. De berørte øjne har ofte hornhindens opacitet eller hævede øjenlåg. Huden er tyk og grov med svær psykomotorisk eller mental retardering i centralnervesystemet.
Diagnose & sygdomsforløb
Den første mistænkte diagnose af I-celle sygdom kan stilles ved visuel diagnose baseret på anamnese. En biokemisk bestemmelse af den lysosomale enzymaktivitet i serumet kan anvendes til at bekræfte diagnosen. Denne bestemmelse afslører et absurd forhold mellem intra- og ekstracellulær aktivitet.
Aktiviteten af phosphotransferase i fibroblasterne kan også bestemmes for at bekræfte diagnosen. Inklusioner svarer til enten mucopolysaccharider, lipider eller oligosaccharider. Molekylær genetisk diagnostik kan fjerne enhver resterende tvivl. Hvis der er en passende historie, kan sygdommen også diagnosticeres som en del af prenatal diagnose.
På grund af den lave udbredelse anbefales faktisk prænatal oparbejdning kun, hvis der er en familieholdning. Sygdomsforløbet afhænger af symptomerne i det enkelte tilfælde og er ikke direkte forudsigelig. De fleste patienter overlever imidlertid knap ti år. Imidlertid er mildere udviklingsformer ikke helt udelukket i individuelle tilfælde.
Komplikationer
I-celle sygdom kan føre til forskellige komplikationer og klager. Disse genkendes imidlertid sent, så jeg-cellesygdommen kun kan diagnosticeres sent. Symptomerne er relativt inkonsekvente, hvilket ofte gør behandlingen vanskelig. Dette fører normalt til ubehag og misdannelser i hud, øjne og indre organer.
I værste tilfælde kan den berørte person blinde eller dø direkte af organsvigt. Desuden er der en udtalt kort statur og også hjerteproblemer. Øjenlågene er ofte hævede, og der er nedsat intelligens og mental retardering. Det er ikke ualmindeligt, at den berørte person er afhængig af hjælp fra andre mennesker i hverdagen på grund af forsinkelsen for at klare det.
Patientens livskvalitet reduceres kraftigt af I-cellesygdommen. Som regel er der ingen særlige komplikationer i behandlingen af sygdommen. Der anvendes medicin og psykologiske behandlinger, der kan lindre symptomerne. Imidlertid er en komplet og kausal behandling af denne sygdom ikke mulig. Levealderen reduceres af sygdommen.
Hvornår skal du gå til lægen?
I-celle sygdom diagnosticeres normalt umiddelbart efter, at barnet er født. Hvorvidt yderligere behandlingstiltag er nødvendige afhænger af symptomernes type og sværhedsgrad. Mindre misdannelser behøver ikke nødvendigvis at blive behandlet. Klubfødder og deformiteter i ryghvirvlerne er derimod alvorlige misdannelser, der skal behandles kirurgisk og med medicin. Forældre bør omgående konsultere en specialist, hvis den læge, der er ansvarlig på barselhospitalet, ikke allerede har gjort det.
Hvis der opstår en ulykke eller et fald som følge af klagerne, skal barnet føres til hospitalet, eller forældrene skal straks tilkalde alarmtjenesten. I tilfælde af alvorlige misdannelser, som også kan påvirke barnets psyke senere i livet, skal en terapeut konsulteres for at ledsage den medicinske behandling. I-celle sygdommen kræver derfor altid en medicinsk undersøgelse. Den rigtige kontaktperson er børnelæge eller specialist i arvelige sygdomme. I tilfælde af synsforstyrrelser skal en øjenlæge konsulteres.
Læger & terapeuter i dit område
Behandling og terapi
I-celle sygdommen betragtes som uhelbredelig. En kausal terapi findes derfor ikke. Behandling er kun symptomatisk og understøttende. Psykoterapeutisk pleje af berørte familier udgør en stor del af understøttende terapi. Symptomatisk behandling afhænger af det enkelte tilfælde. Knoglesymptomerne behandles ofte ved at give bisphosphonater.
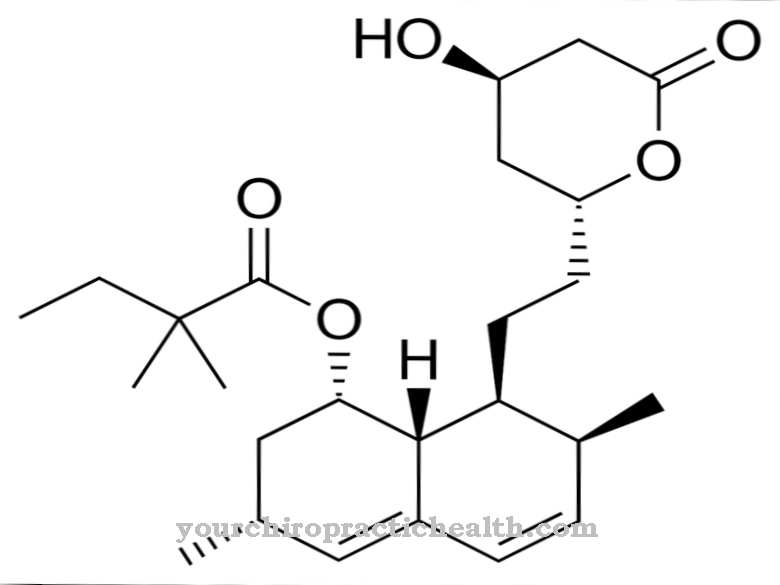
Disse medikamenter er kendt fra behandlingen af osteoporose og har en høj affinitet for knogleoverfladen. Især i regionen med resorptionslakkerne fastgør de sig til knoglerne. Dermed hæmmer de de knoglededbrydende osteoklaster og reducerer på denne måde knogleresorption. Lægemidlerne er pyrophosphatanaloger med en carbonholdig P-O-P-binding.
En enzymatisk hydrolyse finder ikke sted på dem. Aminobisphosphonaterne er blandt de seneste af disse stoffer. Derudover er alendronat, clodronat, etidronat, ibandronat, pamidronat og risendronat godkendt i Tyskland fra den samme lægemiddelgruppe. Det samme gælder tiludronat og zoledronat.
Ud over disse lægemidler kan knoglemarvstransplantationer også bruges til behandling af I-celle sygdomme. Succesen med denne behandling har kun været begrænset i tidligere tilfælde. Genterapier undersøges nu som en ny terapeutisk tilgang til gendefekter. Genterapier har vist indledende succes i dyremodeller. Indtil videre har de ikke været i stand til at blive brugt i praksis på mennesker. Imidlertid vil dette forhold antagelig ændre sig i fremtiden.
Du kan finde din medicin her
➔ Medicin mod smerterOutlook og prognose
I-celle sygdom er en arvelig sygdom, der endnu ikke er behandlet symptomatisk. Prognosen er følgelig negativ. Selvom symptomerne kan reduceres markant ved tidlig behandling, tager I-cellesygdommen næsten altid et alvorligt forløb.
Den korte statur og skader på de indre organer og hoved reducerer allerede forventet levealder betydeligt. Desuden kan misdannelser i ansigt, hud og øjne reducere forventet levetid, men først og fremmest også den berørte persons livskvalitet. Nogle af de berørte når 40-50 år, men de fleste af dem dør i barndom eller ungdom.
Hvis I-cellesygdommen ikke behandles, dør de syge ofte i de første leveår. Prognosen er derfor temmelig negativ. Ikke desto mindre gives udsigten til et relativt symptomfrit liv, hvis patienten behandles som en del af en omfattende terapi og om nødvendigt placeres i en institution for fysisk handicappede. Fysioterapi og terapeutiske foranstaltninger kan forbedre patientens velbefindende markant på lang sigt.
forebyggelse
I-celle sygdom kan kun forhindres ved en molekylær genetisk test inden familieplanlægning. Som en del af den prænatal diagnose kan forventede forældre også beslutte at afslutte graviditeten.
Efterbehandling
I de fleste tilfælde har dem, der er ramt af I-celle sygdom, ingen eller meget få opfølgende foranstaltninger til rådighed. Sygdommen skal anerkendes af en læge så tidligt som muligt, så en yderligere forværring af symptomerne kan forhindres. Da dette er en genetisk bestemt sygdom, bør en genetisk undersøgelse og konsultation altid udføres først i tilfælde af et ønske om at få børn for at undgå arv af I-cellesygdommen til efterkommere.
De fleste patienter er afhængige af forskellige medicin mod denne sygdom. Det er vigtigt at sikre, at doseringen er korrekt, og at medicinen tages regelmæssigt. Hvis noget er uklart, der er bivirkninger, eller hvis du har spørgsmål, skal en læge altid konsulteres først.
Ligeledes har mange syge behov for psykologisk støtte med denne sygdom, selvom kærlige diskussioner med forældre eller pårørende kan have en positiv effekt på sygdomsforløbet. En påvirket person har brug for hjælp og støtte i hverdagen fra deres egen familie. I mange tilfælde begrænser eller reducerer I-cellesygdommen den forventede levetid væsentligt.
Du kan gøre det selv
Patienter, der lider af I-cellesygdom, kan ty til forskellige konservative og alternative behandlingsmetoder. Konservativ terapi fokuserer på at lindre symptomer og lidelser.
Brug af hjælpemidler såsom krykker eller ortopediske indlægssåler kan bremse forløbet af de respektive misdannelser og således også reducere smerterne. Medicin hjælper med at lindre smerter og kan suppleres med alternative tiltag såsom massage eller akupunktur. Alternative behandlinger skal drøftes med den ansvarlige læge på forhånd. Lægen kan muligvis henvise patienten direkte til en homøopat eller give yderligere tip til, hvordan man behandler det respektive symptom.
Da I-celle sygdommen normalt er dødelig på trods af alle behandlingsmuligheder, bør der søges terapeutisk rådgivning. Ikke kun de berørte skal arbejde gennem deres frygt. Slægtninge og venner har normalt brug for støtte til håndtering af sygdommen og dens mulige negative resultat. Deltagelse i en selvhjælpsgruppe er også en mulighed for patienten og deres pårørende. Kontakt med andre syge hjælper med at acceptere sygdommen, og ofte kan andre syge også foreslå yderligere behandlingsforanstaltninger og strategier for det daglige liv med I-cellesygdom.

.jpg)
.jpg)
.jpg)



.jpg)



.jpg)