Det Hippel-Lindau syndrom er en arvelig godartet tumorsygdom primært af nethinden og lillehjernen. Det er baseret på en misdannelse af blodkarene. Andre organer kan også blive påvirket.
Hvad er Hippel-Lindau syndrom?

© deagreez - stock.adobe.com
Det Hippel-Lindau syndrom er en meget sjælden godartet, klumpet vævsændring hovedsageligt i området nethinden og lillehjernen. Såkaldte angiomas (blodsvampe) forekommer som tumorer. Derfor kaldes sygdommen ofte retinocerebellær angiomatose udpeget. Hjernestammen og rygmarven påvirkes også ofte.
Tumorerne udvikler sig fra præformer af bindevævet og består af blodkar. De er for det meste godartede, men kan også degenerere ondsindet. Undertiden findes tumorer i bugspytkirtlen, binyrerne, epididymis eller nyrerne. Især nyretumorer kan udvikle sig til kræft oftere. Sygdommen blev opkaldt efter den tyske øjenlæge Eugen von Hippel og den svenske patolog Arvid Lindau. I 1904 opdagede von Hippel angiomas i nethinden.
22 år senere, i 1926, beskrev Arvid Lindau angiomas i rygmarven. Ud over navnet Hippel-Lindau syndrom er sygdommen også under Von-Hippel-Lindau-Czermak syndrom, Hippel-Lindau sygdom, retinocerebellær angiomatose eller Retinal angiomatose kendt. Det er et neurokutant syndrom, der er kendetegnet ved vaskulære misdannelser i forskellige organer. Neurokutane syndromer er sygdomme, der manifesterer sig i huden og centralnervesystemet.
årsager
Hippel-Lindau-syndromet er genetisk. Sygdommen er arvet som en autosomal dominerende egenskab. Imidlertid afhænger sværhedsgraden af syndromet også af mange andre faktorer. Selvom den genetiske defekt overføres til den næste generation, varierer sygdommens sværhedsgrad i familien. En spontan mutation forekommer hos 50 procent. Så halvdelen af de syge i familien har ingen arvelige problemer.
Dette betyder dog, at flere mutationer i HL-genet på kromosom 3 kan være ansvarlige for udviklingen af sygdommen. Dette gen har en stor indflydelse på udviklingen af blodkar og cellecyklussen. Det er ikke kun en mutation i HL-genet, der fører til dysregulering af blodkarene. Det er nu blevet konstateret, at når sygdommen bryder ud, er der et antal mutationer, der er fordelt over hele genet.
Symptomer, lidelser og tegn
Hippel-Lindau syndrom manifesterer sig generelt i udmattelse, højt blodtryk og hovedpine. Derudover er der neurologiske symptomer såsom balanceforstyrrelser, forstyrrelser i bevægelseskoordination eller tegn på intrakranielt tryk. Synsforstyrrelser (synsforstyrrelser) er ofte de første symptomer. Symptomerne er dog stort set afhængige af tumorenes størrelse og placering.
Angiomas findes normalt i nethinden, i dele af centralnervesystemet, i rygmarven eller i hjernestammen. Hjernen er sjældent påvirket. Undersøgelser afslører ofte misdannelser i andre organer også. Cyster findes især i bugspytkirtlen, leveren eller nyrerne. Godartede arteriovenøse vaskulære misdannelser er også til stede i leveren. Når binyrerne har en angiom, udvikles et pheochromocytoma.
Der er former for Hippel-Lindau syndrom med og uden pheochromocytoma. Pheochromocytoma er en godartet tumor i binyrerne, der producerer en forøget mængde af hormonerne adrenalin og noradrenalin. Puls og blodtryk øges. Blodtrykket stiger med intervaller og reagerer især stærkt i stressende situationer.
Diagnose & sygdomsforløb
Påvisningen af flere hæmangiomer i nethinden i øjnene er et klart bevis på Hippel-Lindau-syndromet. En familiehistorie giver information om enhver ophobning i familien eller pårørende. Billeddannelsesprocedurer kan påvise mulige tumorer i nyrerne, binyrerne, bugspytkirtlen eller leveren.
Komplikationer
Forskellige organer kan blive påvirket af Hippel-Lindau syndrom. I de fleste tilfælde lider den pågældende dog af en generel sygdomstilstand. Dette fører til en alvorlig hovedpine, og den berørte person ser udmattet ud. Desuden opstår højt blodtryk, som i værste fald kan føre til et hjerteanfald. Patienten klager også over nedsat syn og begrænset mobilitet.
Patientens koordinering kan også forstyrres af Hippel-Lindau syndrom. I mange tilfælde har Hippel-Lindau-syndromet også en negativ effekt på patientens opførsel, så det ikke er ualmindeligt, at de bliver dygtige. Pulsen stiger, selv i enkle og lette situationer, så stressede situationer kan føre til sved eller panikanfald for den pågældende. Uden behandling af Hippel-Lindau syndrom reduceres forventet levealder normalt.
Syndromet kan ikke behandles i alle tilfælde. Dette gælder især, hvis syndromet er genetisk. Symptomatisk behandling kan imidlertid begrænse symptomerne og muligvis fjerne tumoren. Det nøjagtige forløb af sygdommen afhænger af sværhedsgraden af svulsten. Patientens forventede levetid kan derfor også være begrænset.
Hvornår skal du gå til lægen?
I tilfælde af en generel sygdomstilfælde, ubehag eller træthed, skal der foretages tæt selvobservation. Hvis træthed fortsætter på trods af en afslappende nattesøvn, er dette et tegn på et sundhedsmæssigt problem. Hvis symptomerne vedvarer i flere uger, er der en grund til en kontrol. Hvis symptomerne spreder sig, eller hvis de intensiveres, skal en læge konsulteres. Kontakt en læge i tilfælde af forhøjet blodtryk, nedsat koordination eller motoriske bevægelsessekvenser.
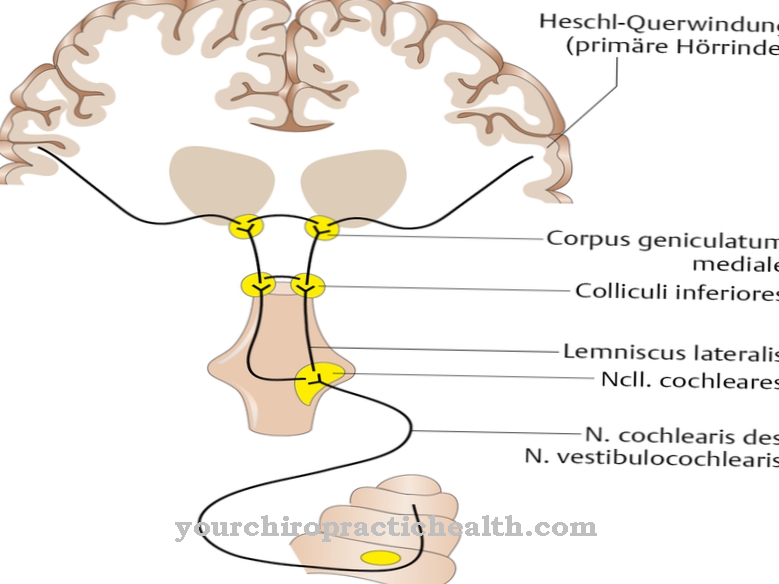
Hvis blodtrykket stiger usædvanligt, især i stressede livssituationer, er et lægebesøg nødvendigt. Hvis der er en følelse af tryk inde i hovedet, hovedpine, synsforstyrrelser eller en begrænsning af det auditive system, er der grund til bekymring. I alvorlige tilfælde er der fuldstændigt høretab. Dette skal undersøges og behandles så hurtigt som muligt. Problemer i ryggen, et fald i fysisk ydeevne og en diffus smerteoplevelse bør afklares af en læge.
Hvis symptomerne ikke vises uden grund, har den pågældende brug for en lægeundersøgelse. I tilfælde af funktionelle forstyrrelser i mave-tarmområdet, oplevelse af varme eller humørsvingninger, anbefales en konsultation med en læge. Ubehag i nyreområdet eller unormale forhold ved urinering betragtes som advarsler fra kroppen, der skal følges op.
Læger & terapeuter i dit område
Behandling og terapi
Da Hippel-Lindau-syndrom er en genetisk sygdom, er en årsagsbehandling ikke mulig. Imidlertid kan eksisterende angiomas fjernes ved en række forskellige procedurer. Disse inkluderer laserkogulation, kryoterapi, brachyterapi, transpupillær termoterapi, fotodynamisk terapi, strålebehandling, protonterapi eller medikamentel behandling. Ved laserterapi denatureres mindre angiomas ved lokal overophedning. Det syge væv dør og heles på dette tidspunkt.
Kryoterapi bruger temperaturer så lave som minus 80 grader til at fryse perifere angiomas i nethinden. I brachyterapi bruges radioaktiv stråling til at ødelægge angiomas. Transpupillær termoterapi kan anvendes til retinoblastomer, choroidale melanomer eller choroidale hemangiomer. Det fungerer på grundlag af opvarmning af tumoren med infrarød stråling. I tilfælde af angiomas er præsentationen af behandlingssucces selvmodsigende. Nogle undersøgelser har rapporteret om succes med behandling af angiom. I andre undersøgelser var behandlingen ineffektiv.
I fotodynamisk terapi bruges lys i kombination med et lysaktivt stof. Verteporfin bruges som et let aktivt stof i aktuelle undersøgelser. En forbedring i synet bemærkes. Makulært ødem kan dog forekomme. Strålebehandling viser ikke nogen signifikante resultater. Synet kan forbedres, men ikke alle tumorer krymper jævnt. De bedste resultater opnås med små angiomas. Protonterapi fungerer med meget høj præcision. Det bruges, når angiomene er nær følsomt væv.
Outlook og prognose
Prognosen for Hippel-Lindau syndrom afhænger i vid udstrækning af typen og placering af de forskellige tumorer. Den gennemsnitlige levealder blev bestemt til at være 50 år. Imidlertid kan forventet levealder og livskvalitet øges markant ved tidlig påvisning og behandling af tumorer. Selvom tumorer oprindeligt simpelthen deformerer bindevævet i blodkarene og er godartede, kan nogle af dem omdanne til ondartede tumorer. Nyrecarcinomer, som er den vigtigste årsag til den høje dødelighed, udvikles derefter særligt ofte.
Pankreatiske carcinomer (bugspytkirtelkræft) og carcinomer i andre organer forekommer også. Kræft i bugspytkirtlen er en af de særligt aggressive tumorer, der hurtigt kan føre til død. En anden almindelig dødsårsag er hæmangioblastom i hjernen, hvilket kan føre til hjerneblødning. Generelle symptomer såsom højt blodtryk, hovedpine og træthed samt neurologiske symptomer afhænger også af de enkelte tumorer.
Nogle patienter har ikke symptomer, hvis de ikke allerede har haft hæmangioblastomer i centralnervesystemet. Hemangioblastomer i nethinden kan føre til problemer med nethinden, efterhånden som sygdommen skrider frem og kan endda føre til fuldstændig blindhed. Endvidere er tumorer, der forårsager fuldstændigt høretab, mulige hos ca. 10 procent af patienterne. Sygdomsforløbet kan være så forskelligt, at en nøjagtig prognose for den berørte ikke er mulig.
forebyggelse
Der er ingen forebyggelse af Hippel-Lindau syndrom, fordi det er en genetisk sygdom. Hvis der er en familiær ophobning af sygdommen, bør genetisk rådgivning udføres, hvis barnet ønsker at få børn.
Efterbehandling
Efter at have fået diagnosen Hippel-Lindau-syndrom, ændrer livet sig for dem, der er berørt. Fra nu af skal du følge din krop nøje og konsultere en læge for at afklare hver nye klump. Jo tidligere masser der behandles, jo større er sandsynligheden for en vellykket behandling, og jo færre er komplikationerne.
Patienter skal have regelmæssige kontrol hele livet. Da nye rumlige krav kan vises i hele kroppen, skal disse udføres af læger fra forskellige specialiteter. Ved årlige generelle kliniske undersøgelser diskuteres alle følbare masser, blodtrykket måles, og yderligere terapi diskuteres. Årlige oftalmologiske undersøgelser muliggør tidlig påvisning af retinal hæmangioblastomer.
Indsamlingsurin kontrolleres også årligt, hvilket kan bruges til at diagnosticere pheochromocytomer. MR-undersøgelser af hoved og rygmarv udføres hvert tredje år for at afbilde og behandle rygmarvshæmioblastomer. En MR af maven tjener også til at udelukke pheochromocytomer, nyrecellekarcinomer og bugspytkirtelsvulster.
De behandlende læger kan også bruge andre undersøgelsesmetoder, såsom positronemissionstomografi eller enkeltfotonmissionstomografi eller scintigrafi for at supplere diagnosen af tumors type og spredning. I individuelle tilfælde kan en kateterundersøgelse af blodkar være nødvendig for at udslette karene, der fører til tumoren, og for at lette efterfølgende behandling.
Du kan gøre det selv
Med Hippel-Lindau-syndrom er der ingen særlige muligheder for selvhjælp til rådighed for de berørte. Desværre kan syndromet ikke forhindres eller behandles med årsag, så der gives kun symptomatisk behandling. Selv efter vellykket behandling er patienten imidlertid ofte afhængig af regelmæssige undersøgelser for at diagnosticere og behandle andre tumorer på et tidligt tidspunkt.
Tumorerne fjernes normalt ved operation. Typen af intervention afhænger stærkt af tumorens placering og sværhedsgraden. Som regel reduceres imidlertid ikke den berørte persons levealder af Hippel-Lindau-syndrom, da tumorer kan fjernes og er godartede. Ofte kan psykologiske forstyrrelser eller depression undgås ved at tale med andre berørte mennesker eller med nære venner og familie. Især med børn bør en informativ samtale altid finde sted for at informere dem om de mulige konsekvenser af sygdommen.
I de fleste tilfælde kan behandling af Hippel-Lindau syndrom også forbedre den berørte persons syn. Disse er dog stadig afhængige af visuelle hjælpemidler for at klare hverdagen. På grund af det høje blodtryk, bør stressede situationer og anstrengende sportsgrene eller aktiviteter undgås. Dette beskytter patientens cirkulation og hjerte.
.jpg)

.jpg)



.jpg)



.jpg)