Det Crigler-Najjar syndrom er en ekstremt sjælden arvelig sygdom. Hos patienter med denne sygdom forstyrres blodmetabolismen på grund af en reduceret aktivitet af enzymet UDP-glucuronyltransferase. Terapimulighederne spænder fra fototerapi til levertransplantation.
Hvad er Crigler-Najjar syndrom?

© Alila Medical Media - stock.adobe.com
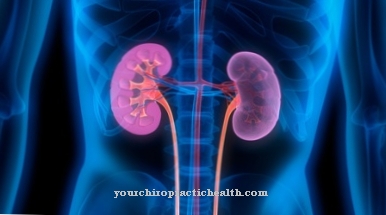
Lægen beskriver en medfødt og ekstremt sjælden sygdom i hæmoglobinmetabolismen som Crigler-Najjar syndrom. Bilirubin er et affaldsprodukt af hæmoglobin og er 90 procent albuminbundet i serum hos sunde mennesker og er primært til stede. Ukonjugerede bilburinniveauer stiger hos patienter med Crigler-Najjar-syndrom. Aktiviteten af det bilirubin-behandlende UDP-glucuronyltransferaseenzym er alvorligt begrænset på grund af en mutation.
Frem for alt viser dette patologiske virkninger på leveren. Udbredelsen af denne sygdom er angivet i et forhold på en til en million. Sygdommen er opkaldt efter lægerne John Fielding Crigler og Victor Assad Najjar. Du beskriver syndromet for første gang i det 20. århundrede. I henhold til moderne medicinsk viden kan der skilles mellem to former for Crigler-Najjar syndrom, der benævnes CN type I og CN type II.
årsager
Crigler-Najjar syndrom er forårsaget af en genetisk defekt. I henhold til aktuel forskning er både den første og den anden type af sygdommen baseret på en defekt i UGT1-genet, som er placeret på kromosom to. Den første type er specifikt en mutation af eksoner to til fem af det tilsvarende gen. Denne type sygdom arves som en autosomal recessiv egenskab.
Dette betyder, at to bærere af den ubrudte genetiske defekt har den samme chance for et sundt barn som for et sygt barn. For at mutationen skal videreføres, skal begge forældre i det mindste være bærere af den defekte allel. Type to af Crigler-Najjar-syndrom overføres på den anden side i en autosomal dominerende arveform. Med denne arv er et defekt gen tilstrækkeligt til arv.
Du kan finde din medicin her
➔ Medicin mod gulsot og leverproblemerSymptomer, lidelser og tegn
Begge former for Crigler-Najjar-syndrom er kendetegnet ved, hvad der kaldes gulsot. Huden, slimhinderne og de indre organer i patienten bliver gul på grund af det udegraderede bilirubin. Ud over behandlingen af bilirubin forstyrres også behandlingen af medikamenter og steroidhormoner. Alle andre leverværdier for patienterne er inden for det normale interval. I den første type syndrom er enzymaktiviteten nul eller stærkt reduceret. I denne form forekommer gulsot umiddelbart efter fødslen.
Bilirubin metaboliseres næppe, og kun små mængder udskilles i afføringen. Selv administration af UDP-glucuronyltransferaseenzym kan ikke reducere stigningen i bilirubin i plasma. Type II af syndromet er noget mildere. Den resterende aktivitet af enzymet er omkring ti procent, og induktionen af enzymet UDP-glucuronyltransferase kan reducere stigningen i bilirubin i plasma.
Diagnose & kursus
Lægen diagnosticerer normalt Crigler-Najjar syndrom ved at tage lever- og blodprøver. Sygdomsforløbet adskiller sig med typen. I type I antager medicin generelt et ugunstigt kursus. I denne form af sygdommen opbevares det overflødige bilirubin i centralnervesystemet. Bilirubin encephalopati udvikler sig.
I medicin betyder dette indtrængning af bilirubin i nervefibre og ind i hjernen. Alvorlige neurologiske mangler opstår som en del af dette fænomen. De fleste patienter af type 1 dør derfor i barndommen. Hos type II-patienter er prognosen temmelig gunstig. I de fleste tilfælde angriber gulsot i deres tilfælde kun huden. Dette kan reducere livskvaliteten på grund af vedvarende kløe. For tidlig død er imidlertid ikke at forvente.
Komplikationer
Neurologiske komplikationer er ikke ualmindelige ved en arvelig sygdom, der påvirker leveren, såsom Crigler-Najjar syndrom type 1. I denne svære form af leversygdom er postnatalt gulsot som følge af hyperbilirubinæmi den værste komplikation. Med den mildere sygdomsform, det såkaldte Arias-syndrom, er alvorlige komplikationer mindre almindelige.
Gulsot kan også manifestere sig her. Det er imidlertid mildere på grund af de stadig aktive enzymrester. Som et resultat kan han behandles bedre. Ikke desto mindre er livskvaliteten altid begrænset af Crigler-Najjar syndrom.
I det mest alvorlige tilfælde af Crigler-Najjar syndrom type 1 skal den nyfødte patient behandles øjeblikkeligt. Behandling af postpartum-komplikationer hos personer med denne type Crigler-Najjar-syndrom kan kræve en levertransplantation i den tidlige barndom. Inden dette bliver nødvendigt, prøver de behandlende læger at forhindre yderligere følgevirkninger af Crigler-Najjar syndrom med konservative behandlingsmetoder.
Eventuelle neurologiske følger skal undertrykkes eller i det mindste udsættes. Hvis dette ikke fungerer, er en levertransplantation uundgåelig. Denne operation medfører store risici hos nyfødte. I hvilket omfang den allogene transplantation af leverceller viser sig at være nyttig og livreddende ved Crigler-Najjar syndrom af type 1 er endnu ikke undersøgt tilstrækkeligt. Hvis phenobarbital administreres dagligt i Crigler-Najjar syndrom type 2, kan sygdomsrelaterede komplikationer undgås.
Hvornår skal du gå til lægen?
Hvis du har symptomer på gulsot eller mærkbar svær kløe, skal du kontakte en læge. Den mærkbare misfarvning af huden indikerer en sygdom, der skal afklares og behandles om nødvendigt. Lægen kan afgøre, om dette er Crigler-Najjar-syndrom, der er baseret på en anamnese og lever- og blodværdier. Senest, når komplikationer bliver mærkbare, indikeres et øjeblikkeligt lægebesøg. Det er bedst at gå direkte til børnelæge med børn, der viser neurologiske abnormiteter.
Akutlægen skal straks advares i tilfælde af hjerte-kar-klager og svigtssymptomer, såsom cirkulationscirkulation eller koma. Crigler-Najjar-syndrom ses normalt i den tidlige barndom. Forældre, der selv lider af en arvelig sygdom eller har tilfælde af syndromet i deres nærmeste familie eller pårørende, bør omgående konsultere en læge, hvis symptomerne nævnes. Ud over den praktiserende læge er andre kontakter også specialister i arvelige sygdomme, neurologer eller internister. Hvis barnet bliver berørt, skal der søges psykologisk støtte på et tidligt tidspunkt.
Læger & terapeuter i dit område
Behandling og terapi
Foranstaltningerne til behandling af Crigler-Najjar syndrom er forskellige alt efter type. Hos type II-patienter anbefaler lægen normalt administration af epilepsimedicinen fenobarbital. Denne gave finder sted en gang om dagen for livet. Lægemidlet siges at stimulere enzymaktivitet og dermed sænke bilirubin-koncentrationen i plasmaet på en sikker måde. For patienter af type 1 består terapi normalt af tre forskellige søjler.
Normalt deltager de i en såkaldt blålys terapi en gang om dagen. Denne behandling gør bilirubin vandopløseligt. Lægemiddeladministrationen af tinprotoporphyrin er beregnet til at reducere stigningen i bilirubin, der nu er vandopløseligt. Lægemidlet er en hæmmer af hemeoxygenase. Denne heme-oxygenase er et enzym, der nedbryder heme til jern. Disse to terapeutiske foranstaltninger afrundes sædvanligvis ved indgivelse af calciumcarbonat og calciumphosphat. Målet er at frigive bilirubin fra organismen til tarmen.
Dette stimulerer udskillelsen af det overflødige stof. Denne tredelte terapi forlænger patienternes forventede levetid. De forventede komplikationer i det neurologiske område kan i det mindste blive forsinket med disse terapeutiske foranstaltninger. Under visse omstændigheder kan en levertransplantation også være nyttig for patienter af den første type. Denne transplantation bør ideelt set udføres relativt tidligt.
Terapeutiske fremgangsmåder til stamceller testes stadig i denne sammenhæng. En kausal behandling og dermed udsigten til en kur mod Crigler-Najjar syndrom eksisterer endnu ikke. Fremskridt inden for genterapi kan ændre det i den nærmeste fremtid.
Outlook og prognose
Det videre forløb af Crigler-Najjar-syndrom afhænger meget af dets nøjagtige sværhedsgrad, så en generel forudsigelse ikke er mulig.
I mange tilfælde er de epileptiske anfald begrænset ved hjælp af medicin. Derudover er mange syge afhængige af fototerapi for at begrænse symptomerne. Behandling af syndromet øger patientens forventede levetid markant, men kan ikke helbrede det fuldstændigt. I alvorlige tilfælde afhænger de berørte af en levertransplantation for at fortsætte med at overleve. I mange tilfælde skal dette gøres i en ung alder.
Hvis Crigler-Najjar-syndrom ikke behandles, fører det normalt til den pågældende død eller til en markant nedsat levealder. En kausal behandling af syndromet er ikke mulig, så kun symptomerne kan begrænses. Forældre skal gennemgå genetisk rådgivning, hvis de vil have børn igen for at forhindre, at syndromet gentager sig hos et barn. Da mange af de kirurgiske indgreb skal finde sted umiddelbart efter fødslen, er de forbundet med høje risici.
Du kan finde din medicin her
➔ Medicin mod gulsot og leverproblemerforebyggelse
Crigler-Najjar syndrom er en genmutation. Derfor kan sygdommen ikke forhindres. Som en del af en DNA-sekvensanalyse kan par dog have sandsynligheden for, at et sygt barn vurderes i familieplanlægningen.
Efterbehandling
I tilfælde af Crigler-Najjar-syndrom er der generelt få opfølgningsforanstaltninger tilgængelige for dem, der er berørt. I tilfælde af denne sygdom er den pågældende primært afhængig af en hurtig og frem for alt tidlig diagnose, så der ikke forekommer yderligere komplikationer, og symptomerne på sygdommen ikke fortsætter med at forværres. Da Crigler-Najjar-syndrom er en arvelig sygdom, kan det heller ikke helbredes fuldstændigt, så patienten er afhængig af livslang terapi.
Hvis du vil have børn, anbefales genetisk rådgivning stærkt for at forhindre gentagelse af denne sygdom hos dine efterkommere. Selvhelende kan ikke forekomme i dette syndrom. Da behandlingen ofte udføres ved hjælp af medicin, skal patienten altid sikre sig, at den tages regelmæssigt med den rigtige dosering. Regelmæssige undersøgelser fra en læge er også meget vigtige for permanent at overvåge symptomerne.
I alvorlige tilfælde af sygdommen kan en levertransplantation imidlertid også være nødvendig, så levealderen ikke sjældent reduceres som et resultat af Crigler-Najjar syndrom. Kærlig pleje og støtte til ens familie er meget vigtig for at forhindre depression eller andre psykologiske forstyrrelser.
Du kan gøre det selv
Patienter med Crigler-Najjar syndrom er alvorligt nedsat i hverdagen. Mens type CN II-syge normalt kun skal overholde medicinen, bestemmer fototerapi især dagligdagen hos CN I-patienter.
At sove ubehandlet for maksimal lyseksponering fører til, at syge mennesker ofte fryser om natten i den kolde sæson. Om sommeren kan den termiske stråling fra det blå lysapparat forstyrre søvnen. Moderne enheder med LED-belysning og markant reduceret varmegenerering giver et middel. Generelt skal der sikres fleksibel klimaanlæg i soveværelset, og beskyttelsesbrillernes pasform kontrolleres. Tab af vand og salt som følge af øget fordampning på grund af lysenergien skal kompenseres.
Andre aktiviteter er stærkt begrænset af flere timers fototerapi. På grund af størrelsen og den manglende transportabilitet af terapienhederne er ferieture også vanskelige at implementere. Imidlertid er bærbare enheder nu tilgængelige. I individuelle tilfælde kan de berørte også behandles ved hjælp af lysoptiske lysmåtter.
For syge børn og unge fører det ydre udseende af Crigler-Najjar syndrom ofte til problemer i kontakt med andre børn. Forældre og pårørende lider ikke bare af drilleri osv. Du er også belastet af behovet for regelmæssig kontrol under fototerapi om natten.


.jpg)



.jpg)



.jpg)