Det Kolesterolopbevaringssygdom er en lysosomal opbevaringssygdom og medfødt metabolisk sygdom med genetisk basis. Sygdommen er arvelig og er forårsaget af en genetisk mutation i de gener, der koder for lysosomal syrelipase. Den symptomatiske behandling af patienten foregår konservativt med medicinering eller gennem terapeutiske trin til enzymerstatning.
Hvad er kolesterolesteropbevaringssygdom?

© 4. livsfotografering - stock.adobe.com
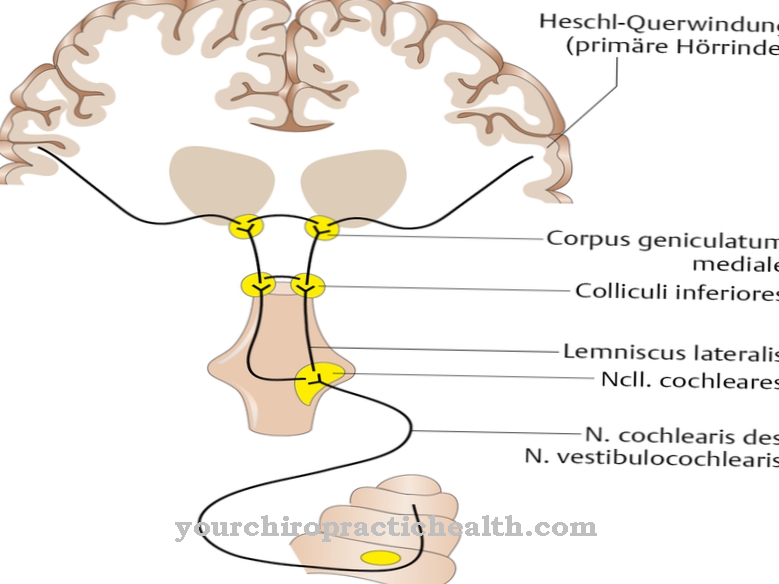
Gruppen af lysosomale opbevaringssygdomme omfatter et antal medfødte sygdomme, der kan spores tilbage til utilstrækkelig eller mangelfuld aktivitet af de såkaldte lysosomer. Alle sygdomme i gruppen er metaboliske sygdomme. En sådan sygdom er kolesterolesterlagringssygdommen, der ofte kaldes CESD er forkortet.
Som alle lysosomale opbevaringssygdomme er CESD også baseret på en mangel på aktivitet af visse lysosomer. Manglen på aktivitet i denne sygdom angår den lysosomale syrelipase, der nedbryder kolesterolestere og triacylglycerid hos raske mennesker. Cholerestinesteroplagringssygdom er en ekstremt sjælden metabolisk sygdom og er forbundet med arvelighed. I denne sammenhæng svarer arv til autosomal recessiv arv.
Sygdommen er en af de medfødte metaboliske sygdomme. De første symptomer behøver dog ikke manifestere sig umiddelbart efter fødslen. Den primære årsag til sygdommen er en genetisk mutation. En sygdom med en lignende genmutation er Wolman sygdom. I modsætning til denne sygdom er kolesterolesterlagringssygdommen kendetegnet ved et meget mildere forløb, da en resterende aktivitet af den lysosomale syrelipase opretholdes, og kolesterolesternedbrydningen kan føre til mindst uden for leveren.
årsager
Patienter med cholerestinesteroplagringssygdom lider af en enzymdefekt i lysosomal syrelipase. Denne defekt fører til en reduceret nedbrydning af cholerestestere. Dette resulterer i en ophobning af kolesterolestere og de lige så lidt nedbrydede triglycerider. Det kodende gen for sur lipase er placeret i DNA'et på kromosom 10 i genlokuset q23.2 til 23.3. Ti eksoner udgør genet.
Årsagen til choleretinesterlagringssygdom er en vrøvl eller missense-mutation i de involverede eksoner. Rammeskift eller springing af visse eksoner kan også være årsagen. Det muterede genprodukt viser reduceret aktivitet, således at næsten ingen lipider af lysosomet kan trænge ind i cytoplasmaet. På denne måde afbrydes kontrolsløjfen til regulering af intracellulære kolesterolkoncentrationer.
En intracellulær lav kolesterolkoncentration opstår og bringer den endogene kolesterolsyntese og LDL-receptoraktiviteten til en opregulering. Som et resultat optager lysosomet endocytoseret kolesterol. På grund af den endogene syntese af kolesterol overbelastes cellerne, og der dannes lipidvakuoler. Vakuolerne fører til et tab af funktion af de enkelte celler, udløser fibrose og forårsager celledød.
Du kan finde din medicin her
➔ Medicin mod smerterSymptomer, lidelser og tegn
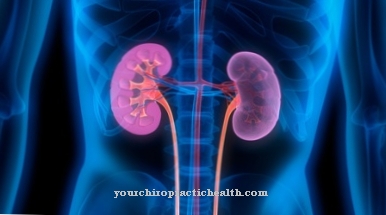
Cholerestinesteroplagringssygdom er kendetegnet ved et karakteristisk kompleks af kliniske symptomer. Lever abnormiteter er især typiske. Som et organ er leveren markant involveret i metabolismen af kolesterol, hvilket forklarer den foretrukne første manifestation af sygdommen i leveren. Hos patienter med cholerestinesterlagersygdom manifesterer symptomerne sig i de fleste tilfælde meget senere end hos patienter med Wolman-sygdom.
I sidstnævnte manifesterer de første symptomer sig normalt mere eller mindre umiddelbart efter fødslen. I modsætning hertil kan patienter med cholerestinesterlagersygdom forblive asymptomatiske i lang tid. Det vigtigste symptom er hepatomegali. Leveren svulmer op og udvikler sig til en fed lever, når aflejringerne skrider frem. Derudover forekommer hypercholesterolæmi i de fleste tilfælde.
Denne hypercholesterolæmi svarer til en lipidmetabolismeforstyrrelse på grund af øget kolesterolniveau i blodet. Disse symptomer er normalt forbundet med hyperlipidæmi og en nedsat HDL-koncentration. Ud over hævelse i leveren og funktionsnedsættelse kan de fleste af symptomerne udelukkende diagnosticeres ved laboratoriediagnostik.
diagnose
Til diagnose af cholerestinesterlagringssygdom kræves en laboratorieblodanalyse, der kan detektere det ændrede lipidmønster og dokumentere eventuelle skumceller, der forekommer. Derudover kan en leverbiopsi udføres som en del af diagnosen, der viser massive lysosmale akkumuleringer.
Med hensyn til differentiel diagnose skal sygdommen differentieres fra andre lysosomale opbevaringssygdomme inden for rammerne af diagnosen. Denne sondring foretages normalt i sammenhæng med enzymatiske aktivitetstest. De genetiske tests til identifikation af mutationen bruges sjældent til at differentiere.
Komplikationer
Kolesterolesteroplagringssygdom kan føre til forskellige klager og komplikationer, der primært afhænger af sygdommens sværhedsgrad. I de fleste tilfælde påvirkes imidlertid leveren. Hos mange patienter vises de første tegn på kolesterolesterlagringssygdom umiddelbart efter fødslen, så for eksempel leveren er hævet, eller hvis den senere udvikler sig til en såkaldt fedtlever.
Oftest oplever patienter også smerter og stikkende fra hævelsen. Diagnosen er normalt relativt let med en blodprøve, så der er ingen forsinkelse i diagnosen. Desværre er en kausal terapi og behandling af kolesterolesterlagringssygdommen ikke mulig, så først og fremmest symptomerne skal begrænses.
Dette resulterer i en reduceret absorption af kolesterol. Patienten er dog afhængig af at tage medicin resten af sit liv. Ellers forårsager kolesterolesterlagringssygdommen ikke yderligere klager eller komplikationer. Levealderen reduceres heller ikke med terapi. Patientens hverdag er sjældent begrænset af sygdommen. Når man planlægger børn, skal man dog undersøge sandsynligheden for at tilbe sygdommen.
Hvornår skal du gå til lægen?
Kolesterolesteroplagringssygdom diagnosticeres normalt umiddelbart efter fødslen. Typiske tegn, der peger på sygdommen, og som skal afklares og behandles, er en hævet lever, og som sygdommen skrider frem, tegn på fedtlever. Hvis barnet klager over skarp smerte i leveren, skal børnelægen omgående konsulteres. Dette gælder især, hvis der er yderligere klager, der indikerer kolesterolesterlagringssygdommen.
I tilfælde af alvorlige komplikationer skal den akutmedicinske service altid kontaktes. Selvom livstruende symptomer sjældent forekommer med sygdommen, kan kronisk leversygdom udvikles. Derfor skal du tale med en læge ved de første tegn på kolesterolesterlagringssygdom.
Hvis der allerede er kendte tilfælde af sygdommen i familien, anbefales en undersøgelse umiddelbart efter fødslen. Under visse omstændigheder kan sygdommen også diagnosticeres prenatal. Lægen vil derefter diskutere med forældrene, hvilke yderligere foranstaltninger der er mulige som en del af en behandling.
Læger & terapeuter i dit område
Behandling og terapi
Cholerestinesteroplagringssygdommen skyldes en genetisk defekt. Derfor er der ingen hittil kausale terapeutiske trin til behandling af patienterne. En kausal terapi ville kun være mulig i forbindelse med genterapimetoder. Genterapi er endnu ikke nået den kliniske fase. Derfor betragtes lagringssygdommen stadig i dag som uhelbredelig og behandles kun symptomatisk. Symptomatisk behandling fokuserer på en reduceret optagelse af kolesterol.
Til dette formål inhiberes patientens tarmcholesterolabsorption konservativt med medicin. Lægemidler såsom cholestyramin og ezetimibe er egnede til inhibering. Derudover får patienter normalt statiner, som hæmmer HMG-CoA-reduktase. I den seneste tid er der også etableret nye behandlingsmuligheder, især enzymerstatningsterapier.
For patienter med cholerestinesterlagersygdom spiller enzymet sebelipase alfa en vigtig rolle.Enzymerstatningsterapier med dette enzym anvendes i øjeblikket i kliniske forsøg og blev godkendt og vurderet positivt af Det Europæiske Lægemiddelagentur sidste år.
Outlook og prognose
Kolesterolesterlagringssygdom har en ugunstig prognose. Sygdommen betragtes som uhelbredelig og kan føre til vanskelige komplikationer. Den recessive arvelige sygdom behandles symptomatisk af læger, da der af juridiske årsager i øjeblikket ikke er tilladelse til indblanding i human genetik.
Alvorligheden af sygdommen er individuel og derfor forskellig for hver patient. Følgelig findes der heller ikke nogen ensartet behandlingsplan. Når leveren er kompromitteret, falder udsigterne til forbedring af helbredet dramatisk. Lever hævelse eller fedt leversygdom kan føre til andre sygdomme. Der er ofte mere betændelse eller skrumpelever i leveren. På dette tidspunkt er det næsten umuligt at lindre symptomerne. Den syge person trues med leversvigt og dermed for tidlig død.
Patienter, hvis lever ikke lider skade, har et betydeligt bedre helbred. Så længe de følger lægenes retningslinjer og undgår indtagelse af produkter, der indeholder kolesterol ud over medicin, oplever de en god livskvalitet på trods af opbevaringssygdommen.
Da behandlingen er en langtidsbehandling, forværres sundhedstilstanden inden for kort tid, så snart lægemidlet er ophørt. En usund livsstil har også en øjeblikkelig negativ indflydelse på patientens velbefindende.
Du kan finde din medicin her
➔ Medicin mod smerterforebyggelse
Cholerestinesteroplagringssygdom er en genetisk sygdom. Da der ikke vides at være eksterne faktorer som årsagsfaktorer, kan sygdommen kun og kun forhindres gennem genetisk rådgivning i familieplanlægningsfasen. Imidlertid kan dette heller ikke udelukke nye mutationer.
Efterbehandling
I de fleste tilfælde er der ingen særlige opfølgende foranstaltninger til rådighed for dem, der er berørt med kolesterolesterlagringssygdom. Patienten er primært afhængig af en hurtig diagnose, så symptomerne kan afhjælpes korrekt, da dette ikke kan føre til selvheling. Derfor skal den berørte person kontakte en læge, så snart de første symptomer og klager optræder for at forhindre, at symptomerne forværres.
Da dette er en genetisk sygdom, bør der altid foretages en genetisk undersøgelse og rådgivning, hvis du vil have børn for at forhindre, at kolesterolesterlagringssygdommen opstår igen. I de fleste tilfælde behandles sygdommen med medicin.
Den berørte person skal sikre sig, at de tages regelmæssigt, og at doseringen er korrekt, så symptomerne lindres permanent. Kontakt først en læge, hvis noget er uklart, eller hvis du har spørgsmål. Med kolesterolesteroplagringssygdom er mange patienter afhængige af familie og familiehjælp. Kontakt med andre mennesker, der er berørt af sygdommen, kan også være meget nyttig.
Du kan gøre det selv
Kolesterolesterlagringssygdom er en genetisk lidelse. Derfor er der i øjeblikket hverken konventionelle eller alternative metoder til at behandle årsagen til sygdommen. Følgelig er der ingen tilgængelige selvhjælpsforanstaltninger, der bekæmper sygdommen kausalt.
Patienter kan dog stadig yde et vigtigt bidrag til at undgå progression af sygdommen og alvorlig følgeskade, især i leveren. En diæt med lavt kolesteroltal er af central betydning. Patienter bør indhente omfattende og kompetent information om de specielle diætkrav for kolesterolesterlagringssygdommen.
Den behandlende læge er ikke altid den bedst mulige kontakt, da ernæringsspørgsmål stadig næppe spiller en rolle i uddannelsen af læger. De berørte bør derfor konsultere en ernæringsfysiolog eller en økotrofolog.
Generelt findes kolesterol kun i fødevarer af animalsk oprindelse. Skift til en vegansk diæt giver derfor mening for de berørte. Under alle omstændigheder bør fødevarer af animalsk oprindelse, som er særlig højt i kolesterol, undgås.
Disse indbefatter især fedtkød, pølseprodukter, slagteaffald, æg, smør, fløde og sødmælk. Æg spises ofte skjult. Patienter er ofte ikke klar over, at mange typer pasta, især pasta og kager, såvel som færdigretter og mayonnaise, indeholder store mængder æg og tilsvarende høje mængder kolesterol.




.jpg)



.jpg)