Det Allan-Herndon-Dudley-syndrom er en mutation i SLC16A2-genet, der ændrer skjoldbruskkirtelhormonentransportøren MCT8 og forårsager forringet iodothyroninoptagelse i muskelvæv og centralnervesystemet. På grund af mutationen lider de ramte muskelsvaghed samt forsinkelser i mobil og mental udvikling. AHDS er uhelbredelig og er hidtil kun blevet behandlet med indgivelse af triiodothyroacetat.
Hvad er Allan - Herndon - Dudley syndrom?

Forsinkelser i den fysiske, mentale eller følelsesmæssige udvikling hos unge og børn sammenfattes som udviklingsforsinkelser eller forsinkelse. Udviklingsforsinkelser kan have forskellige årsager. For eksempel kan udløseren til den forsinkede udvikling ligge i det centrale nervesystem.
Dette er f.eks. Tilfældet med Allan - Herndon - Dudley Syndrome (AHDS). Ud over alvorlig mental retardering er syndromet kendetegnet ved forstyrrelser i motorisk udvikling. Den første beskrivelse af syndromet går tilbage til 1944. Det kliniske billede, du beskriver, er en arvelig sygdom, dvs. en genetisk lidelse.
Sygdommen påvirker mandlige spædbørn i størstedelen af alle til dato dokumenterede tilfælde. Udviklingsforstyrrelser og deres konsekvenser er i næsten alle tilfælde klart synlige fra fødslen. AHDS er en ekstremt sjælden sygdom. Derfor har undersøgelsen af Allan-Herndon-Dudley syndrom hidtil været dårlig.
årsager
AHDS er en arvelig genetisk lidelse forårsaget af en mutation i SLC16A2-genet. Dette er det kodende gen for den såkaldte thyroidhormontransportør MCT8. Denne transportør formidler optagelsen af iodothyroniner i muskel- og nervevæv.
På grund af mutationen forekommer der forstyrrelser i optagelsen af skjoldbruskkirtelhormoner, der forstyrrer centralnervesystemet og dermed forringer udviklingen af nervesystemets celler. Muskelvæv og hjerne bliver fattige på grund af den mutationsrelaterede dysregulering af det aktive skjoldbruskkirtelhormon, som de faktisk er afhængige af.
Syndromet overføres som en X-bundet recessiv arv. Kvinder kan arve sygdommen, men på grund af deres dobbelt X-kromosomstruktur bliver de sjældent syge. Syge mænd er ikke i stand til at formere sig.
Selvom mutationen er genetisk, ud over denne interne faktor, spiller eksterne faktorer sandsynligvis en rolle i begyndelsen af sygdommen. På grund af sjældenheden og det begrænsede forskningsgrundlag er rollen for disse eksterne faktorer endnu ikke blevet afklaret endnu.
Du kan finde din medicin her
➔ Medicin mod muskelsvaghedSymptomer, lidelser og tegn
Allan-Herndon-Dudley syndrom er en medfødt sygdom, der normalt manifesterer sig i småbørn eller babyer. De berørte lider af mere eller mindre alvorlig muskelsvaghed. Børnenes muskelvæv er mærkbart underudviklet. Muskels svaghed ledsages snart af leddeformiteter.
Kontraktioner er også hyppigt ledsagende symptomer. Børnenes mobilitet forringes i stigende grad af kontrakturer og deformiteter. Af denne grund forekommer de berørte ofte unaturligt statisk eller endda bevægelsesfri. På grund af den mutationsrelaterede underforsyning af skjoldbruskkirtelhormoner lider de berørte ofte også muskelkramper eller foretager ufrivillige bevægelser med arme og ben.
Ofte kan de berørte ikke bevæge sig uafhængigt. I de fleste tilfælde er de motoriske svækkelser forbundet med alvorlige psykiske lidelser. For eksempel er størstedelen af patienterne ikke i stand til at tale. I individuelle tilfælde kan AHDS karakteriseres af mange andre symptomer inden for mental og fysisk udvikling.
Diagnose & kursus
Lægen mistænker normalt først AHDS i anamnese. I laboratorietest indikerer et forhøjet T3-niveau med normale FT4- og TSH-niveauer Allan - Herndon - Dudley syndrom. Afbildning af det centrale nervesystem er normalt en del af diagnosen.
Muskelsvagheder på grund af motoriske neuronale sygdomme kan udelukkes fra den differentierede diagnose. Prognosen for patienter med Allan-Herndon-Dudley syndrom er relativt dårlig. Indtil videre er sygdommen uhelbredelig. Undersøgelser har antydet, at tidspunktet for diagnosen kan spille en kritisk rolle i patientens prognose.
Komplikationer
Som alle kromosonalt nedarvede lidelser, kan Allan - Herndon - Dudley syndrom ikke behandles helbredende. Den mest almindelige bivirkning af Allan - Herndon - Dudley syndrom - udtalt muskelsvaghed - kan behandles med fysioterapi. En sådan behandling, der sigter mod at styrke muskler, kan være smertefuld for patienten.
Især små børn nægter ofte behandling på grund af smerterne. På trods af intensiv træning fører fysioterapi ikke altid til den ønskede succes. Det svarer til taleterapistøtten fra Allan - Herndon - Dudley-patienten. Selvom reduktionen i sprogfærdigheden kan forbedres med intensiv træning, fører behandlingen ikke altid til succes på grund af den mest høje grad af mental svækkelse af de berørte.
Patientens frustration og en stor belastning for hele familien er en af de mest alvorlige komplikationer i behandlingen af Allan - Herndon - Dudley syndrom. Muskelkramper og bevægelser af ekstremiteterne, som ikke kan påvirkes, kan behandles med indgivelse af muskelafslappende midler. Komplikationer kan ses i de til tider alvorlige bivirkninger af medicinen.
Træthed, en generel udmattelsesfølelse og ubehag skal nævnes ud over stress på mave-tarmkanalen. Langvarig brug af relanxanter skader også leveren og nyrerne. Hvis Allan - Herndon - Dudley syndrom ikke behandles, vil de berørte ikke være i stand til at gøre nogen væsentlig fremgang med hensyn til deres mentale eller motoriske evner.
Hvornår skal du gå til lægen?
I mange tilfælde er direkte behandling af Allan-Herndon-Dudley syndrom ikke mulig. Af denne grund er behandlingen hovedsageligt symptomatisk og er rettet mod de individuelle klager og forsinkelser. Som regel skal forældre derefter se en læge, hvis barnet har muskelsvaghed.
Dette kan mærkes ved udmattelse eller vedvarende træthed. Medicinsk rådgivning er også nødvendig, hvis Allan-Herndon-Dudley syndrom bremser mental og motorisk udvikling.
Hvis behandlingen ikke finder sted i barndommen, kan det føre til betydeligt ubehag og begrænsninger i voksen alder. En læge skal konsulteres, især hvis patienten ikke længere kan tale. Behandling er også nødvendig for muskelkramper. Hvis det er en akut nødsituation, kan du gå direkte til hospitalet eller ringe til en ambulance.
I de fleste tilfælde vil Allan-Herndon-Dudley-syndrom blive behandlet af en læge eller en børnelæge. Imidlertid skal de enkelte klager undersøges og behandles af den respektive specialist eller terapeut.
Læger & terapeuter i dit område
Behandling og terapi
AHDS er en årsagsmæssig ubehandlet sygdom. Da der ikke er nogen tilgængelige behandlingsformer til korrigering af den primære årsag, er sygdommen hidtil ikke blevet helbredelig. I mellemtiden antyder fremskridt inden for genterapi, at genterapimetoder snart vil blive godkendt til daglig klinisk praksis.
I hvilket omfang patienter med syndromet ville drage fordel af godkendelse er endnu ikke afklaret. På nuværende tidspunkt er der ingen etableret eller standardiseret behandlingsmulighed for patienter med AHDS inden for området symptomatisk terapi. For nogle få år siden betragtede forskere administrationen af TRIAC som en mulig symptomatisk terapimulighed.
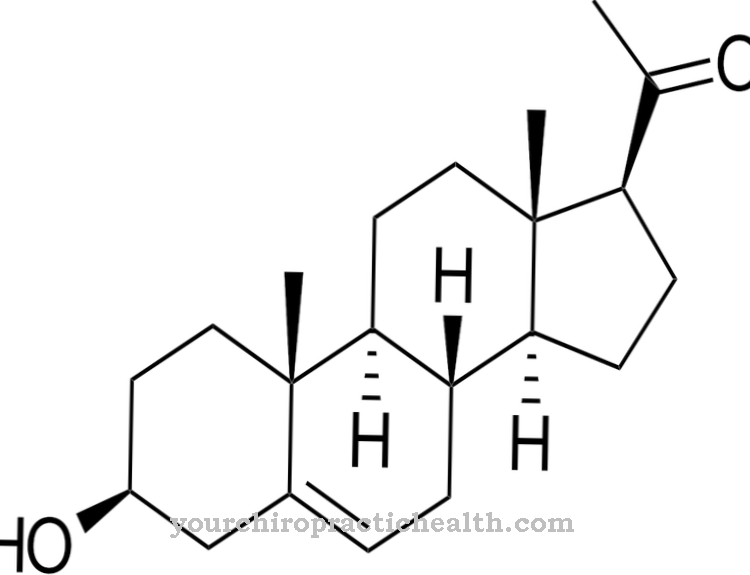
TRIAC er et ikke-klassisk thyroidhormon, triiodothyroacetat. Indgivelsen af hormonet blev udført i en klinisk undersøgelse af berørte børn, men kunne ikke opnå synlige resultater. Resultaterne af undersøgelsen er ikke nødvendigvis meningsfulde, fordi administrationen af hormonet begyndte relativt sent.
Af denne grund blev TRIAC i 2014 stadig betragtet som den bedst mulige terapi. I et tilfælde i 2014 blev der dokumenteret en betydelig forbedring i motorisk og mental udvikling under behandling med TRIAC. Terapien blev startet på den berørte person i den tidlige spædbarn.
Resultaterne af de hidtidige undersøgelser indikerer således, at tidspunktet, hvor behandlingen startes, har en effekt på terapiresultaterne, som ikke bør undervurderes for patienter med AHDS. Ledsagende understøttende terapier såsom ergoterapi og fysioterapi eller tidlig indgriben kan teoretisk bruges til at forbedre livskvaliteten og patienternes færdigheder. Der er imidlertid næppe noget bevis på effektiviteten af en sådan tilgang i forbindelse med AHDS-patienter.
Outlook og prognose
Allan-Herndon-Dudley-syndrom forårsager en række forskellige symptomer hos de fleste patienter. Først og fremmest lider de ramte muskelsvaghed. Dette betyder, at normale aktiviteter eller sportsgrene ikke længere let kan udføres for den pågældende. Der er også store forsinkelser i intellektuel og mobil udvikling. Patientens koncentration er klart begrænset og reduceret.
Der er stadig stærke kramper i musklerne og dermed ofte ufrivillige bevægelser eller rykninger. Efterhånden som Allan - Herndon - Dudley syndrom skrider frem, kan de berørte ikke længere tale. Patientens hverdag er betydeligt begrænset af syndromet, og livskvaliteten reduceres. I nogle tilfælde er patienterne derefter afhængige af hjælp fra andre mennesker i deres hverdag.
Det er normalt ikke muligt at behandle Allan - Herndon - Dudley syndrom med årsag. Af denne grund er behandlingen kun symptomatisk. De berørte er afhængige af forskellige behandlingsformer, som imidlertid ikke altid fører til et positivt forløb af sygdommen. I nogle tilfælde begrænser Allan - Herndon - Dudley syndrom forventet levealder for de berørte.
Du kan finde din medicin her
➔ Medicin mod muskelsvaghedforebyggelse
AHDS kan kun forhindres gennem genetisk rådgivning. Mutationsbærere kan for eksempel beslutte imod at få deres egne børn.
Efterbehandling
Behovet for opfølgning af genetisk forårsaget Allan - Herndon - Dudley syndrom påvirker kun mandlige spædbørn. Problemet er, at der ikke er nogen passende form for terapi for denne arvelige tilstand. De alvorlige konsekvenser af defekter i transmissionen af skjoldbruskkirtelhormon kan næppe forbedres. Forsøg på at give de berørte børn lettelse ved at give dem specielle skjoldbruskkirtelhormoner har fejlet.
Problemet er, at sygdomsgrundlaget normalt allerede er etableret i moders krop. De beskadiger det ufødte barn permanent. Fra dette synspunkt begynder behandlingen for sent, nemlig først efter fødslen. I efterbehandlingen kan kun de allerede tilstedeværende skader behandles. Der er dog håb. I 2014 blev der kendt en sag, hvor et spædbarn med Allan-Herndon-Dudley syndrom blev behandlet med TRIAC. Opfølgningspleje var stadig nødvendigt, fordi barnet ikke kunne helbredes. I det mindste hans symptomer blev lindret.
Allan-Herndon-Dudley-syndrom er delvis forårsaget af en mangelfuld blod-hjerne-barriere. Dette antyder undersøgelser på Cedars Sinai Hospital. Skjoldbruskkirtelhormonerne kan ikke fungere på grund af den defekte blod-hjerne-barriere. Dette gælder også skjoldbruskkirtelhormoner, der indgives efter barnet er født. Bioteknologi eller genetisk forskning kan muligvis hjælpe. Alle forsøg på behandling mislykkes i øjeblikket. Dette påvirker også opfølgningen af hårdt beskadigede børn.
Du kan gøre det selv
Allan-Herndon-Dudley syndrom er en alvorlig tilstand, der endnu ikke er behandlet effektivt. Forældre kan dog stadig tage nogle skridt til at støtte terapi.
For det første er regelmæssig kognitiv træning og fysisk aktivitet vigtig. En omfattende terapi, der afhængigt af sværhedsgraden af syndromet kan være sammensat af tale- og læsetræning, men også generelle hjerneøvelser, tilbyder også fysioterapiøvelser. Træningen skal være individuelt tilpasset symptomerne. Forældrene til de berørte børn bør derfor sikre sig, at foranstaltningerne vælges optimalt, og at barnet ikke bliver overvældet.
I tilfælde af alvorlige psykiske lidelser kan barnet muligvis have permanent støtte i hverdagen. En ambulant pleje kan være en vigtig lettelse for forældrene. Inpatientbehandling er lige så vigtig, som kan understøttes derhjemme ved regelmæssig overvågning af symptomer.
Forældre bør også drage fordel af psykologisk rådgivning og om nødvendigt også gå til en selvhjælpsgruppe, fordi kontakt med andre berørte mennesker gør det lettere at håndtere sygdommen. Derudover modtager forældre ofte vigtige tip til, hvordan man håndterer et sygt barn.
.jpg)
.jpg)



.jpg)





.jpg)