Det Smith-Lemli-Opitz syndrom er et kogenital misdannelsessyndrom. Årsagen er en af i alt 70 genmutationer på kromosom 11q13.4. Sygdommen er arvet på en autosomal recessiv måde og er en ekstremt sjælden sygdom med multiple organdeformationer og nedsat kolesterolbiosyntese.
Hvad er Smith-Lemli-Opitz syndrom?

© jijomathai - stock.adobe.com
Det Smith-Lemli-Opitz syndrom falder ind i gruppen af autosomale recessive arvelige misdannelsessyndromer. En genmutation forårsager en metabolisk lidelse i biosyntesen af kolesterol i sygdommen. Syndromet er den mest almindelige cogenitale lidelse ved kolesterolbiosyntese. Udbredelsen af sygdommen i Europa er mellem ca. 1: 60.000 og 1: 10.000.
Sygdommen kan således klassificeres som en sjælden sygdom, skønt den er en af de mest almindelige cogenitale sygdomme i kolesterolbiosyntesen. Syndromet er endnu sjældnere på kontinenterne i Asien og Afrika. Sygdommen blev først beskrevet i 1964. Genetikerne D. W. Smith, L. Lemli og J. Marius Opitz dokumenterede symptomkomplekset fra et videnskabeligt synspunkt. Lidt over 300 sager er rapporteret siden da.
Drenge påvirkes oftere end piger. Symptomerne er sandsynligvis mere udtalt hos piger og derfor vanskeligere at diagnosticere. Sygdommen er medfødt, men udvikler sig gradvist fra fødslen og fremefter og er derfor kendetegnet ved forskellige former. Syndromet er opdelt i typer I og II afhængigt af symptomerne.
årsager
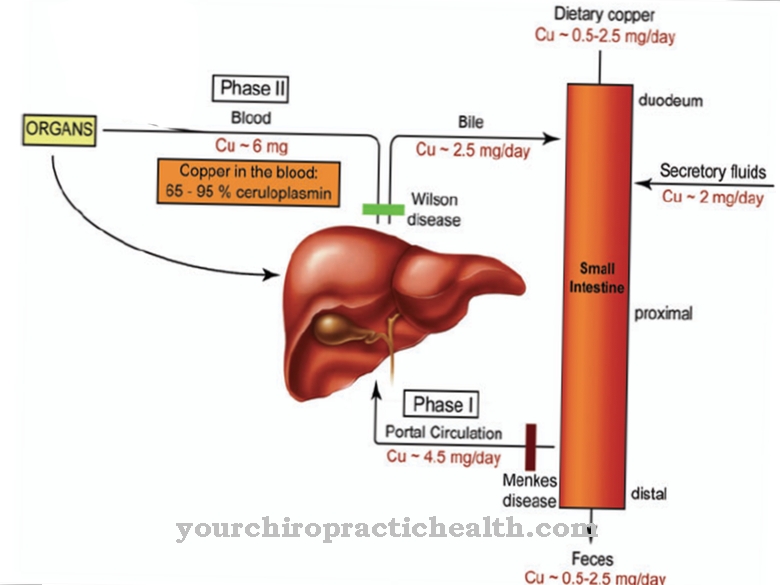
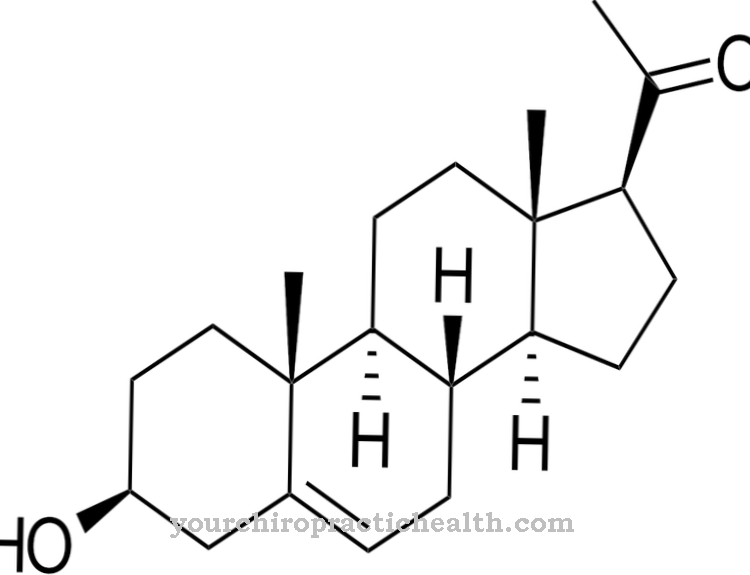
Smith-Lemli-Opitz syndrom er forårsaget af en genmutation, der blev lokaliseret i 1998. Kromosom 11q13.4 betragtes nu som placeringen af mutationen, med mere end 70 forskellige mutationer på dette sted, der hidtil er kendt. Typen af kausal mutation bestemmer sværhedsgraden og typen af symptomer i hvert enkelt tilfælde. Det pågældende gen er 7-sterolreduktasegenet. S. Tint opdagede sammen med sine kolleger, at syndromet forhindrer kroppens egen produktion af kolesterol.
Denne produktion inkluderer omdannelse af forløberen 7-dehydrocholesterol til kroppens eget kolesterol, som ikke kan fungere på grund af en enzymfejl som et resultat af mutationen. Der er derfor et overskud af 7-dehydrocholesterol i kroppen af de berørte. På samme tid er der et generelt underskud i kolesterol. På grund af den autosomale recessive arv af syndromet, skal begge forældre bære det defekte gen og kan kun videregive det til et barn på denne måde. Der er 25 procent sandsynlighed for, at de efterfølgende børn af forældre med et sygt barn også påvirkes af misdannelsessyndromet.
Symptomer, lidelser og tegn
Børn med Smith-Lemli-Opitz-syndrom fødes med typiske craniofacial misdannelser, især mikrocephaly, en fremtrædende pande og en lille næse med en bred næsestamme. Ud over en antervert nare er der mikrogenius. En ganespalte og lukning af linsen observeres også ofte, især grå stær og grå stær.
Blepharoptosis er også til stede. Mentalt og cerebralt udvikles uønsket udvikling over tid, hvilket resulterer i et mentalt handicap. Holoprosencephaly og irritabilitet kan også forme det kliniske billede. Ud over selvskadende adfærd kan syndromet provocere autistisk opførsel. Derudover er der flere organdeformationer, især hjertet og urogenitalkanalen.
Hypospadier og kryptorchidisme er de mest almindelige urogenitale misdannelser. Ud over overskydende fingre eller tæer kan tå syndaktisk være til stede. Muskelhypotoni, slukningsforstyrrelser og gastroøsofageal reflux er også relevant i forbindelse med symptomkomplekset. Intestinal dysmotilitet og pylorstenose er også almindelige. I type II af syndromet findes der pseudo-hermaphroditisme, hvor de ydre kønsorganer er kvindelige, selvom den mandlige karotype er fremherskende.
Diagnose & sygdomsforløb
Som en prænatal diagnose kan ultralydundersøgelser registrere de typiske fysiske egenskaber ved Smith-Lemli-Opitz syndrom allerede før fødslen. Ud over et vækstunderskud kan man bemærke en hjertedefekt eller fraværet af en nyre. I fostervandsundersøgelsen kan mutationsanalysen allerede give et diagnosebekræftende resultat.
Efter fødslen har børnene en karakteristisk ansigtsform og særlige positioner i ekstremiteterne, så den mistænkte diagnose kan stilles ved visuel diagnose, hvis den prænatal diagnose er mislykket. Den genetiske diagnose sikrer mistanken. Med hensyn til differentiel diagnose skal Smith-Lemli-Opitz-syndrom adskilles fra føtalalkoholsyndrom, Pallister Hall-syndrom, Kaufmann-McKusick-syndrom og Cornelia de Lange-syndrom.
Pätau-syndrom, ATR-X-syndrom og C-syndrom, Zellweger-syndrom og hydrolethalus-syndrom skal også overvejes ved den differentielle diagnose. Det samme gælder Oro-Faciales-Digitales-syndromet, Holoprosencephaly-Polydactyly-syndromet og Meckel-syndromet. Børns forventede levetid afhænger af kolesterolkoncentrationen og behandlingsevnen af organdeformationer. Lavt kolesteroltal og de mest alvorlige misdannelser gør et hurtigt fatalt resultat sandsynligt. Børn med høje kolesterolniveauer og let behandlingsmæssige misdannelser har ikke en alvorlig nedsat levealder.
Komplikationer
På grund af Smith-Lemli-Opitz-syndromet lider de berørte af forskellige misdannelser og deformiteter. Disse har en meget negativ effekt på patientens livskvalitet. Især de indre organer påvirkes af misdannelserne, så døden ofte kan forekomme umiddelbart efter fødslen. Desuden lider de fleste patienter af en ganespalte og også af øjenproblemer.
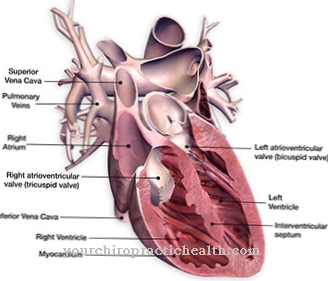
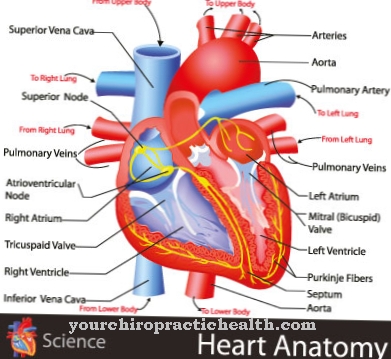
Desuden fører dette syndrom ofte til intellektuel handicap og dermed til intellektuel retardering. De fleste patienter er derfor afhængige af hjælp fra andre mennesker i deres liv og kan ikke længere klare mange hverdagsopgaver på egen hånd. Hjertet påvirkes også af misdannelserne, hvilket kan føre til pludselig hjertedød. Desuden påvirker Smith-Lemli-Opitz syndrom også kønsorganerne, så misdannelser også kan forekomme i disse.
Behandling af Smith-Lemli-Opitz syndrom kan normalt kun være symptomatisk. Der er ingen komplikationer, og nogle af symptomerne kan være begrænset. Imidlertid kan et helt positivt forløb af sygdommen ikke opnås. Det kan ikke forudsiges universelt, om forventet levealder vil være begrænset.
Behandling og terapi
For patienter med Smith-Lemli-Opitz-syndrom er livslang social og medicinsk behandling ofte uundgåelig. Som regel er deres udvikling meget forsinket med hensyn til de kognitive og motoriske områder. I næsten alle tilfælde resulterer dette i en livslang handicap, der ikke tillader en uafhængig livsstil. Derfor ydes først og fremmest støttende behandling.
Som en del af disse tiltag får forældre psykoterapeutisk støtte og ideelt set lære at håndtere deres barns sygdom. Smith-Lemli-Opitz syndrom er uhelbredelig og kan derfor ikke behandles som en årsag. Da der er dokumenteret en forstyrrelse i kolesterolmetabolismen for syndromet, er symptomatisk behandling for at kompensere for kolesterolmangel. Denne behandling udføres ved at give kolesterol.
De mest mange misdannelser i organerne skal korrigeres kirurgisk, så vidt det er muligt. En undtagelse fra dette er den hyppigt dokumenterede multibinding af fingre og tæer, som ikke nødvendigvis kræver kirurgisk indgreb. Samtidige symptomer som synsvansker kan nu også behandles og lindres godt.
Størstedelen af de berørte lider af ernæringsproblemer, såsom sugning og slukning af vanskeligheder eller gastroøsofageal refluks og nedsat gastrisk peristaltik. Derfor skal et gastrisk rør ofte bruges til at sikre et sikkert fødeindtag. Adfærdsproblemer kan muligvis behandles med adfærdsterapi.
forebyggelse
Efter en positiv præenatal diagnose for Smith-Lemli-Opitz syndrom får forældrene mulighed for at afslutte graviditeten. Smith-Lemli-Opitz-syndromet kan kun forhindres på anden måde, hvis par har en molekylær genetisk diagnose udført i deres familieplanlægning, og efter at der er fremlagt bevis for mutationen, træffer beslutning om deres egne børn.
Efterbehandling
Opfølgningsforanstaltningerne for Smith-Lemli-Opitz syndrom (SLOS) er baseret på sværhedsgraden af symptomerne, der forekommer i løbet af sygdommen. I de fleste tilfælde af sygdom har børnene ernæringsproblemer. De klarer sig dårligt. Fokus for opfølgningspleje er derfor først og fremmest en passende ernæring af de berørte børn ved hjælp af nasogastriske flydende fødevarer med højt kalorieindhold og administration af tilstrækkeligt kolesterol.
Det videre sygdomsforløb viser også hos mange af de ramte børn en underudvikling af hjernen. Denne underudvikling fører normalt til fysiske eller psykiske handicap med varierende sværhedsgrad. For eksempel lærer ikke alle berørte børn at gå. For at kompensere for den begrænsede mobilitet tilvejebringes hjælpemidler til hverdagens bevægelse (f.eks. Kørestol, gå- og stående hjælpemidler) som en opfølgende foranstaltning i dette tilfælde.
I tilfælde af mentale symptomer, såsom autoaggression og hyperaktivitet, fortsættes den terapeutisk ordinerede medikamentbehandling som en opfølgende foranstaltning. Derudover udvikler omkring 50 procent af alle berørte børn en moderat til svær hjertefejl, efterhånden som sygdommen skrider frem. Efter operationen af hjertedefekten planlægges elektrokardiografiske (EKG) og sonografiske undersøgelser med regelmæssige intervaller.
For forældre til børn med SLOS anbefales psykologisk konsultation og terapi. Selvstændigt at leve i voksen alder er temmelig usandsynligt. Der forventes omfattende plejeforanstaltninger i opfølgningen af SLOS i voksen alder. Derudover kan organmisdannelser begrænse forventet levealder.
Du kan gøre det selv
Sygdommen er forbundet med adskillige klager, der alvorligt forringer livskvaliteten. Hvis et familiemedlem er diagnosticeret med den genetiske sygdom, skal en læge konsulteres, før barnet er undfanget. Eventuelle risici bør vejes, så der kan træffes forsigtige beslutninger for alle involverede. Derudover skal du deltage i alle check-ups, der tilbydes under graviditet.
Så snart barnets helbredsnedsættelser bemærkes, kan forældre og pårørende tage passende forholdsregler og forberede sig bedre på den fremtidige udvikling. I et stort antal tilfælde er omsorg for nogen med Smith-Lemli-Opitz syndrom en enorm udfordring for de pårørende. Derfor bør de kende og overholde deres fysiske og følelsesmæssige grænser. Det tilrådes at overveje brugen af medicinsk behandling til patienten og parallelt med psykoterapeutisk støtte til de pårørende. Som et resultat kan der ofte opnås forbedringer i behandlingen af sygdommen.
Det skal bemærkes, at stabilitet i det sociale miljø er vigtigt for patienten. Derudover skal du altid være rolig i hverdagen, når der opstår ulykker og udfordringer. Da den pågældende ikke er i stand til at forme sit liv uafhængigt, bør der være en særlig empati med at håndtere ham.
.jpg)
.jpg)
.jpg)





.jpg)





.jpg)