Som Lenz-Majewski syndrom lægen kender en type hyperostotisk kort statur, som er forbundet med en cutix laxa og osteosklerose. Syndromet er baseret på en mutation i PTDSS1-genet på gen locus 8q22.1. En kausal terapi er endnu ikke tilgængelig for de berørte.
Hvad er Lenz Majewski-syndrom?

© deepagopi2011 - stock.adobe.com
Det Lenz-Majewski syndrom er en speciel og ekstremt sjælden form for kort statur. Symptomkomplekset er hidtil kun blevet beskrevet i ni tilfælde. Som et syndrom fra gruppen af hyperostotisk kort statur er komplekset ikke kun kendetegnet ved kort statur, men også af karakteristiske ansigtstræk og en cutis laxa. Progressiv knoglesklerose blev også observeret i de nyligt beskrevne tilfælde.
Progressiv sklerose er også kendt som osteosklerose og svarer til hærdning af knoglevævet. Forekomsten af Lenz-Majewski-syndrom antages kun at være et tilfælde hos en million mennesker. De kliniske udtryk er synonymer til symptomkomplekset Braham-Lenz syndrom og Lenz-Majewski hyperostotisk dværgisme.
Det første synonym går tilbage til den første deskriptor Braham. I sine bemærkninger i det 20. århundrede omtalte han stadig syndromet som Camurati-Engelmann-syndrom. Lidt senere udførte den tyske humangenetiker Lenz og børnelægen Majewski en indledende afgrænsning. Sygdommens navn som Lenz-Majewski-syndrom henviser til denne første afgrænsning mod slutningen af det 20. århundrede.
årsager
Lenz-Majewski syndrom har en genetisk årsag. De hidtil observerede tilfælde ser ikke ud til at have opstået sporadisk, men er underlagt en autosomal dominerende arv. Den gamle alder af fædrene til berørte børn peger især på autosomale dominerende nye mutationer som den primære årsag til symptomerne.
I mellemtiden er det årsagsgen identificeret til trods for de få beskrevne tilfælde. Syndromet er sandsynligvis baseret på en mutation i PTDSS1-genet, som er placeret på genlokuset 8q22.1. Genet koder for et protein kaldet phosphatidylserinsyntase 1.
På grund af mutationen af genet mister proteinet sin funktion, der består i dannelsen af phosphatidylserin. Phosphatidylseriner er vigtige phospholipider, der tælles blandt membrankomponenterne. Denne forbindelse ser ud til at udløse den ekstreme korte statur med patologisk ændrede rygsøjler.
Symptomer, lidelser og tegn
Patienter med Lenz-Majewski-syndrom lider af forskellige kliniske kriterier. Sygdommen manifesterer sig i det tidlige spædbarn og forekommer i løbet af denne tid som en manglende trivsel. Patienter ser ud til at være ældre og progeroid. Dine kraniumsuturer er usædvanligt brede. Det samme gælder hendes fontanel. Ofte umiddelbart efter fødslen bemærkes craniofacial dysmorphism.
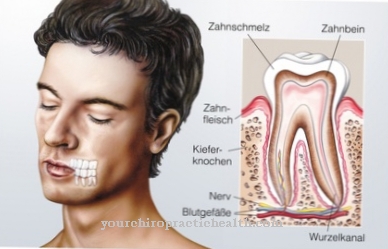
De fleste patienter har en fremtrædende og ekstremt bred pande. Derudover er der ofte hypertelorisme. Ofte blokeres de berørte tårekanaler i deres løb. Auriklerne forekommer for store og mærkbart slappe. Der er normalt defekter i emaljen hos de berørte, som senere fremmer tandfald. De aldrede udseende hos de berørte kan spores tilbage til en såkaldt cutis laxa.
Dette fænomen svarer til tynd og visnet hud, der i stigende grad er kendetegnet ved årer og er forbundet med hernias, kryptorchidisme eller hypospadi. De fleste patienter lider af mental retardering. På fingrene manifesterer syndromet sig som membranøse syndaktylier, der normalt påvirker mellemrummet mellem anden og femte fingre.
Den alvorlige korte statur er det mest karakteristiske symptom på Lenz-Majewski syndrom. Under visse omstændigheder påvirkes lemmerne til den berørte person også af brachydacty og forekommer kraftigt forkortede.
Diagnose & sygdomsforløb
For at stille en første mistænkt diagnose af Lenz-Majewski syndrom har lægen normalt kun brug for det kliniske billede. Derudover kan der arrangeres et røntgenbillede. Billeddannelsen viser typiske kriterier, såsom progressiv osteosklerose på kranietknogler og rygsøjler. Dette tegn kan være forbundet med brede ribben eller kraveben.
Derudover kan der under visse omstændigheder ses diaphyseal sklerose med en udvidelse af knoglerne i røntgenbillede. Hypoplasia af de midterste phalanges taler også for syndromet. Med hensyn til differentiel diagnose skal lægen differentiere syndromet fra symptomkomplekser med et lignende udseende, for eksempel Camurati-Engelmann-syndrom, craniodiaphyseal dysplasi eller craniometaphyseal dysplasi.
En molekylær genetisk test kan udføres for at bekræfte en mistænkt diagnose. Hvis patienten faktisk lider af Lenz-Majewski-syndrom, giver denne analyse bevis for den genetiske mutation.
Komplikationer
Med Lenz-Majewski-syndrom lider de, der er berørt, primært af kort status. Hos børn kan denne klage føre til psykiske problemer eller mobning og drilleri. Livskvaliteten for den berørte person reduceres betydeligt af denne sygdom. Det er endvidere ikke ualmindeligt, at der forekommer tandbrydning og andre defekter.
Uden behandling lider de berørte af alvorlig tandpine og andre ubehagelige symptomer i mundhulen. Det er ikke ualmindeligt, at Lenz-Majewski-syndrom fører til mental retardering, så patienterne er afhængige af hjælp fra andre mennesker i deres hverdag. Ofte lider forældrene og pårørende til de berørte også symptomerne på psykologiske klager eller depression.
Desuden kan individuelle ekstremiteter forkortes som følge af sygdommen, hvilket også kan føre til forskellige restriktioner i livet. Det er ikke muligt at behandle Lenz-Majewski syndrom kausalt. Af denne grund er behandlingen primært rettet mod at reducere de individuelle symptomer. Der er ingen komplikationer, men sygdomsforløbet er ikke helt positivt. Som regel er de, der er berørt af Lenz-Majewski-syndrom, afhængige af hjælp fra andre mennesker i hele deres liv.
Hvornår skal du gå til lægen?
Patienter med Lenz-Majewski-syndrom oplever forskellige symptomer, der skal afklares. Da sygdommen er arvelig, kan symptomer diagnosticeres kort efter fødslen. Forældre bør konsultere en læge nøje, hvis der er symptomer eller komplikationer. Hvis barnet udvikler følelsesmæssige problemer som følge af misdannelser og hudændringer, skal en terapeut indkaldes. Komplikationer på grund af mentale klager skal også afklares af en specialist.
Det er bedst for forældre at holde kontakten med en specialistklinik for genetiske sygdomme. Andre læger skal konsulteres, hvis barnet viser tegn på synsforstyrrelser eller ubehag i munden. Den karakteristiske korte statur skal behandles af en ortopæd kirurg. Hvis Lenz-Majewski-syndromet behandles tidligt, kan symptomerne lettes kraftigt. Derfor er tidlig diagnose nødvendig, selvom symptomerne på hyperostotisk kort statur muligvis endnu ikke er meget udtalt i starten. Efter den første behandling har barnet stadig brug for hjælp fra fysioterapeuter, psykologer og læger.
Behandling og terapi
Indtil videre er der ingen kausale terapeutiske tilgange tilgængelige for patienter med Lenz-Majewski-syndrom. Imidlertid er genterapimetoder til behandling i øjeblikket genstand for medicinsk forskning. Afhængig af udviklingen inden for dette forskningsemne, kan der være fremtidige muligheder for gen-terapi til genteling for syndromet. I øjeblikket skal de berørte dog være tilfredse med symptomatisk behandling.
Denne symptomatiske behandling kan for eksempel omfatte kirurgisk korrektion af misdannelserne. En sådan korrektion behøver ikke altid at finde sted, men er begrænset til syndaktylier, der begrænser patienten i hans hverdag. Den gradvise hærdning af knoglerne kan nedsættes under visse omstændigheder gennem diæt og medicinske indgreb.
Osteosklerose skal overvåges konstant og nøje, så eventuelle brud hurtigt kan behandles. På grund af de mentale og intellektuelle mangler anbefales de berørte patienter normalt tidlig indgriben. Som en del af denne tidlige indgriben kan eventuelle underskud på det intellektuelle plan ideelt reduceres, så de er moderate og næppe bemærkes. Forældrene til de berørte børn bør ideelt få detaljeret rådgivning om mulige finansieringsmuligheder.
Outlook og prognose
Prognosen for Lenz-Majewski syndrom er dårlig. I henhold til den aktuelle medicinske og videnskabelige status er der ingen terapeutisk mulighed, der fører til bedring. Årsagen til sundhedsforstyrrelsen er baseret på en genetisk defekt. Da ændringer i human genetik ikke kan foretages af juridiske grunde, er der ingen måde at korrigere mutationen.
Det videre sygdomsforløb afhænger af sværhedsgraden af de enkelte klager. Ikke desto mindre er livskvaliteten generelt hos en patient med syndromet betydeligt begrænset. Selv hvis symptomerne er milde, er der abnormiteter i vækstprocessen. Kort statur er karakteristisk for sygdommen. Dette kan ikke korrigeres, selv med administration af hormoner. Den visuelle fejl fører til en tilstand af følelsesmæssig nød. Af denne grund øges risikoen for psykologiske komplikationer for de berørte.
I tilfælde af et alvorligt forløb af sygdommen er der ud over de fysiske abnormiteter også kognitive begrænsninger. Den mentale retardering kan allerede understøttes i de første leveår med tidlig støtte. Derefter forbedres den samlede kognitive ydeevne, men der er en mulighed for, at begrænsninger i ydeevneniveauet forbliver for livet. Underskuddene kan minimeres ved hjælp af en terapeut og støtte fra pårørende. Dette gør det muligt for nogle patienter at leve et godt liv.
forebyggelse
Lenz-Majewski-syndrom kan ikke indtil videre forhindres, da symptomkomplekset er forårsaget af en genetisk ny mutation af en grund, der ikke er identificeret.
Efterbehandling
Da Lenz-Majewski syndrom er uhelbredelig, kræves der regelmæssig og omfattende opfølgning. De berørte lider normalt af en række komplikationer og klager, som i værste fald kan føre til den pågældende død. Sygdommen bør derfor genkendes meget hurtigt for at indeholde yderligere klager eller komplikationer.
Lidende skal se en læge regelmæssigt for at gennemgå medicinindstillinger og mulige bivirkninger. Da sygdommen kan være meget stressende for den berørte person og deres pårørende, kan psykologisk støtte anbefales for generelt at lindre lidelsen.
Du kan gøre det selv
Det er ikke muligt at behandle Lenz-Majewski syndrom ved hjælp af selvhjælp. Som regel er heller ikke direkte medicinsk behandling af syndromet mulig, da det er en arvelig sygdom.
Hvis den pågældende har intellektuelle underskud, skal disse afhjælpes gennem intensiv støtte. Jo tidligere denne finansiering begynder, jo større er sandsynligheden for at kompensere for disse underskud. Forældrene kan også gøre forskellige øvelser med barnet i deres eget hjem for at fremme og styrke ånden. Dog bør barnet også få intensiv støtte i børnehaver og skole.
Hærdningen af knoglerne kan lettes ved at tage medicin. Det er vigtigt at sikre, at det tages regelmæssigt, selvom forældre bør være opmærksomme på denne regelmæssighed, især med børn. Hærdningen kan også stoppes gennem en tilpasset diæt. Her skal dog de nøjagtige instruktioner fra lægen om en særlig diæt overholdes. Misdannelserne korrigeres ved kirurgiske indgreb og kan ikke behandles ved hjælp af selvhjælp.
.jpg)











.jpg)

.jpg)