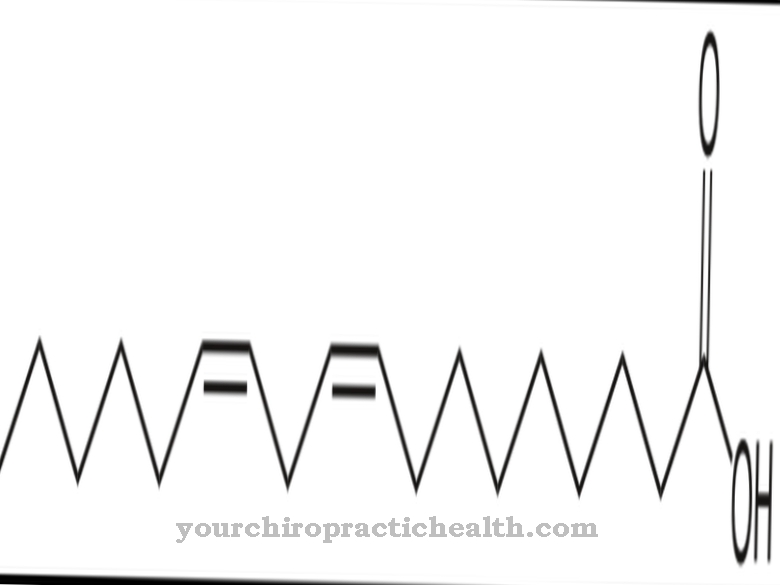
sphingolipider hører til byggestenene i cellemembranen sammen med glycerophospholipider og kolesterol. Kemisk er de afledt af sfingosin, en umættet aminoalkohol med 18 carbonatomer. Hovedsagelig er nervesystemet og hjernen rige på sfingolipider.
Hvad er sfingolipider?
Alle cellemembraner indeholder glycerophospholipider, kolesterol og sfingolipider. Sphingolipider består af bashingstrukturen sphingosin, på hvis aminogruppe esterificeres en fedtsyre. Sphingosin er en aminoalkohol, der indeholder en kæde med 18 carbonatomer. Sphingolipiderne kan opdeles i tre grupper.
Disse er ceramiderne, sfingomyelinerne og glycosfingolipiderne. Ceramider er de enkleste sfingolipider. Her esterificeres sfingosin med en fedtsyre. Der dannes en amfifil dobbeltstruktur med dobbelt lipidlag. Amfifili dannes af de to carbonhydridhaler, der hver peger i den modsatte retning. Sphingomyeliner esterificeres på hydroxylgruppen i sfingosin-skeletet med en fosforsyre, som igen esterificeres enten med en alkohol eller med cholin.
Endelig har glycosfingolipider en glycosidbinding med en sukkerrest på hydroxylgruppen i sfingosin-skeletet. Cerebrosiderne er monohexoser, mens gangliosiderne indeholder en oligosaccharose, der er bundet til glycosid.
Funktion, effekt og opgaver
Sphingolipiderne udfører forskellige funktioner. Deres struktur spiller en vigtig rolle i dette. De enkleste sfingolipider, ceramiderne, er især involveret i strukturen af det liderlige hudlag. På grund af deres amfifilicitet kan de danne et lipid-dobbeltlag, der beskytter huden mod udtørring.
Ud over denne funktion udfører ceramider imidlertid også mange andre opgaver. Disse inkluderer signaloverførsel eller opgaver til styring af celledeling. Foruden glycerophospholipider og kolesterol er sphingomyeliner ansvarlige for cellemembranernes fluiditet og transport af stoffer. Funktionerne af glycosphingolipider er også forskellige. Som allerede nævnt indeholder glycosfingolipiderne en glycosidisk bundet hexose eller et oligosaccharid på hydroxylgruppen i sfingosin-skeletet. Som et resultat har de en hydrofob ceramidkomponent og en hydrofil sukkerbestanddel.
Denne sukkerbestanddel placeres på overfladen af cellemembranen, hvilket kan resultere i celle-celle-interaktioner gennem celleadhæsion. Derfor er de af stor betydning for signaloverførsel af nerveceller. Glycosfingolipider er også stort set ansvarlige for celleinteraktioner i de andre celler.
Uddannelse, forekomst, egenskaber & optimale værdier
Den biokemiske syntese af sfingolipiderne finder sted i det endoplasmatiske retikulum og i Golgi-apparatet. Derfra transporteres de til membranerne ved hjælp af vesikler. Sphingolipiderne konverteres endnu længere i membranerne, så de kan udføre deres mange opgaver der. Alle cellemembraner indeholder sfingolipider. Deres koncentration er imidlertid især høj i hjerneceller og nerveceller. Dette gælder især for gangliosiderne. For eksempel udgør de seks procent af lipiderne i hjernens grå stof.
Cerebrosiderne, der er forestret med en hexose, findes i større udstrækning i hjernen og leveren. I hjernen er det glycosidisk bundne sukker for det meste galactose, mens cerebrosiderne i leveren hovedsageligt indeholder glukose. De enkleste sfingolipider, ceramiderne, findes især i huden og der i stratum corneum (liderligt lag). Som allerede nævnt kan de på grund af deres amfifili danne en barriere der, der beskytter huden mod vandtab. Naturligvis findes ceramiderne også i de andre cellemembraner, da de er nødt til at udføre endnu flere funktioner med hensyn til signaltransmission og cellestyring. Sphingomyelin findes også i alle cellemembraner. Imidlertid er dens højeste densitet igen i neuronerne.
Sygdomme og lidelser
Såkaldte opbevaringssygdomme kan forekomme i forbindelse med sfingolipider. Dette er for det meste genetiske sygdomme, der er kendetegnet ved et manglende eller inaktivt enzym. Som et resultat er nedbrydningen af de tilsvarende sfingolipider ikke længere mulig. Sphingolipidet ophobes i cellen og fører til dets ødelæggelse.
Mange opbevaringssygdomme er dødelige efter mange års lidelse. Typiske lipidopbevaringssygdomme inkluderer Tay-Sachs syndrom og Niemann-Pick sygdom. I løbet af disse sygdomme akkumuleres sfingolipider i cellerne. Tay-Sachs sygdom er forårsaget af en autosomal recessiv arvelig mutation i et gen, der koder for enzymet ß-hexosaminidase A. Dette enzym er ansvarlig for nedbrydning af gangliosid GM2. Som et resultat af dets fiasko ophobes gangliosid GM2 især i nervecellerne. Patienten lider af centralnerves og motoriske lidelser såvel som mental retardering. Denne sygdom fører til død inden for de første tre leveår.
Niemann-Pick sygdom er også arvet som en autosomal recessiv egenskab. Ved denne sygdom akkumuleres sfingomyeliner i cellemembranerne. Endotelceller, mesenchymale og parenchymale celler påvirkes især. Esterificering af kolesterol forstyrres også, så også dette aflejres i cellerne. Der er forskellige former for sygdom i Niemann-Pick sygdom. Disse afhænger af den respektive aktivitet af sfingomyelinasen. I den klassiske form af sygdommen begynder symptomer i løbet af det første leveår. Døden forekommer normalt inden udgangen af det tredje leveår. Hvis sygdommen starter senere, vil symptomerne udvikle sig langsommere. Derefter er Niemann-Pick sygdom karakteriseret ved stigende lever- og miltforstørrelse, kramper, bevægelsesforstyrrelser, muskeltremor og mental retardering.
En anden sphingolipidose er Gauchers sygdom. Gauchers sygdom er Gauchers sygdom. Glucocerebrosider aflejres konstant i forskellige kropsceller, såsom leveren, milten, knoglemarven og makrofager. Denne sygdom fører normalt til død i en alder af to. Gauchers sygdom kan imidlertid behandles ved at erstatte enzymet glucocerebrosidase.







.jpg)

.jpg)


.jpg)
