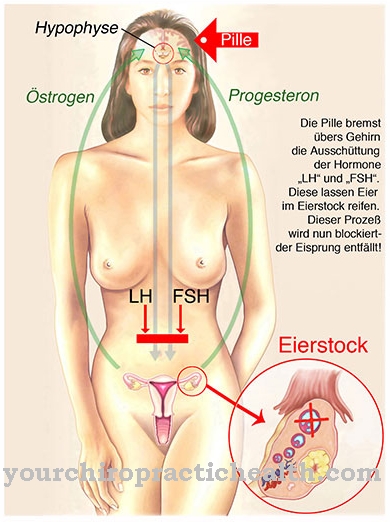
Det Holt-Oram syndrom er et misdannelsessyndrom, der primært er forbundet med hjertefejl og anomalier i tommelfingrene og er forårsaget af en mutation. I de fleste tilfælde forekommer den forårsagende mutation sporadisk og svarer således til en ny mutation. Den kirurgiske korrektion af hjertedefekten er fokus for terapien.
Holt-Oram syndrom?

© SmirkDingo - stock.adobe.com
De medfødte misdannelsessyndromer med overvejende involvering af ekstremiteterne er en gruppe af sygdomme, der inkluderer forskellige deformationer i arme og ben. En af dem er dette Holt-Oram syndrom, også kaldet Hjerte-hånd-syndrom er kendt. Syndromet er forbundet med gruppen af atriodigital dysplasi, som inkluderer medfødte sygdomme med misdannelser i de øvre ekstremiteter og hjertet.
Ud over Holt-Oram-syndrom tilhører hjerte-hånd-syndrom type 2, hjerte-hånd-syndrom type 3 og hjerte-hånd-syndrom af den slovenske type denne gruppe af sygdomme. Holt-Oram-syndromet blev først beskrevet i 1960. Den britiske børnelæge Holt og kardiologen Samuel Oram er de første til at beskrive sygdommen. Holt-Oram-syndromet Dette er en relativt sjælden sygdom og er forbundet med en gennemsnitlig udbredelse af en ramt person i 100.000 mennesker.
Årsagen til misdannelser i hænder og hjerte i Holt-Oram syndrom ligger i genetik. Symptomerne på syndromet er lige så forskellige som dets mistænkte årsager. Misdannelserne i sygdommen koncentrerer sig om hjerte og tommel.
årsager
Selvom Holt-Oram syndrom er en genetisk og medfødt sygdom, er der næppe nogen familiær hyppighed i de hidtil dokumenterede tilfælde. Selvom syndromet ser ud til at være arvet i en autosomal dominerende arveform i individuelle tilfælde, antyder en stor del af sagsdokumentationen dets sporadiske forekomst. Cirka 85 procent af de dokumenterede tilfælde synes at skyldes en ny mutation.
Den primære årsag til Holt-Oram-syndromet ligger i genetiske mutationer ved gen locus 12q23-24.1.Det såkaldte TBX5-gen, der er placeret på kromosom 12 og er et protein involveret i lemmet og lemmet, er lokaliseret på dette genlokus Kodet hjerteudvikling. De nøjagtige funktioner af proteinet er endnu ikke afklaret. Det er heller ikke endnu konstateret, om eksterne faktorer såsom eksponering for toksiner eller underernæring hos moderen under graviditet favoriserer mutationen af TBX5-genet.
Mutationer i genet kan påvises hos mindst op til 70 mennesker ud af 100 patienter med Holt-Oram syndrom. Videnskaben antager imidlertid, at abnormiteter i andre gener også kan forårsage symptomerne på syndromet. For eksempel er misdannelsessyndrom forbundet med treparts tommelfingerpolysyndaktisk.
Symptomer, lidelser og tegn
Patienter med Holt-Osram syndrom lider af et kompleks af misdannelser, der primært påvirker tommelfinger og hjerte. Selvom lokaliseringen af patientens misdannelser er almindelig, er forskellige typer misdannelser på hjerte og tommelfinger mulige. Det kliniske billede er derfor ekstremt varieret.
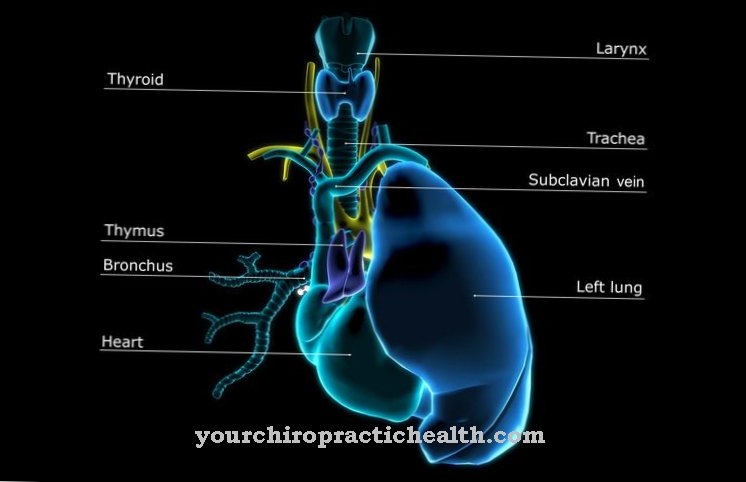
Hjertedefekterne kan manifestere sig, for eksempel i form af en ventrikulær septaldefekt, en atrial septumdefekt, en hjertearytmi eller en ledningsforstyrrelse. Skeletteanomalierne kan svare til reduktionsmisdannelser i tommelfingrene, men afvigelser såsom manglende anvendelse af egerne kan også forekomme.
Mange patienter med syndromet lider også af radioulære synostoser og abnormiteter i ribben, scapula eller clavicle. Derudover er Holt-Oram syndrom forbundet med pectus carinatum og skoliose. Størstedelen af de berørte lider også af syndaktylia i fingre eller tå-falanger. I individuelle tilfælde er disse symptomer forbundet med hypertelorisme.
Diagnose & sygdomsforløb
Holt-Oram syndrom er ofte fejlagtigt diagnosticeret. Ved en differentieret diagnose skal lægen differentiere symptomkomplekset fra Okihiro syndrom efter genmutationer i SALL4-genet på kromosom 20, som er forbundet med de samme armmisdannelser og hjertedefekter. Det, der især er relevant ved den differentielle diagnose, er, at patienter med Okihiro-syndrom normalt har en Duane-anomali.
De myser, er ofte påvirket af nyremissdannelser og har hørselsforstyrrelser, mundanomalier eller øre misdannelser. Trombocytopeni-fravær-radius-syndrom skal også differentieres fra Holt-Oram-syndrom, som hovedsageligt opnås gennem laboratoriediagnostik. Andre kliniske billeder med et klinisk lignende billede er Fanconi-anæmi og Pallister Hall-syndrom.
Levetiden for patienter med Holt-Oram syndrom er ikke under gennemsnittet. Kun i alvorlige tilfælde er der en næppe behandlelig hjertedefekt, der gør prognosen ugunstig.
Komplikationer
Holt-Oram syndrom forårsager en række forskellige misdannelser og deformiteter hos patienten, hvilket kan gøre livet og hverdagen vanskelig. Frem for alt påvirkes hjertet af misdannelserne, så patienten lider af en hjertedefekt. Der er også en hjertearytmi, hvorfra den berørte person kan dø i værste tilfælde.
Anomalier forekommer også på tommelfingrene, så visse bevægelser eller processer i hverdagen også gøres vanskeligere. Det er ikke ualmindeligt, at deformiteter i kroppen fører til drilleri og mobning af andre børn, hvilket kan føre til psykologiske klager og depression hos mange patienter. Det er ikke ualmindeligt, at der forekommer nyresygdomme, som i værste tilfælde kan føre til nyreinsufficiens.
Desuden lider de berørte også af en synshandicap og en hørehæmning. Som regel er forventet levealder uændret som et resultat af Holt-Oram syndrom, så længe der ikke er nogen hjertefejl, der fører til død. En kausal behandling af Holt-Oram syndrom er normalt ikke mulig, så kun symptomerne kan behandles. I mange tilfælde er psykologisk støtte også nødvendig.
Hvornår skal du gå til lægen?
Holt-Oram syndrom diagnosticeres normalt kort efter fødslen. Afhængigt af hvor alvorlige misdannelserne er, kan det berørte barn muligvis have behov for yderligere medicinske undersøgelser. I princippet skal hjertedefekter behandles hurtigt for at reducere risikoen for alvorlige sekundære sygdomme. Hvis der udvikler komplikationer som hjertearytmier eller tegn på atrisk septumdefekt, kræves lægehjælp. En medicinsk professionel bør også konsulteres med abnormiteter i knogletumlen.
Forældrene til de berørte børn skal konsultere deres læge nøje og informere dem om eventuelle usædvanlige symptomer. Da Holt-Oram syndrom er en arvelig sygdom, er det ikke muligt at forårsage kausal behandling. Patienterne kan derfor blive nødt til at blive behandlet i en levetid afhængigt af, hvilke misdannelser der forekommer, og hvad patientens sammensætning er. Da dette ofte også medfører følelsesmæssige klager, vises ledsagende psykologisk støtte. Børn, der lider af mobning eller drilleri, skal søge terapeutisk rådgivning med deres forældre.
Læger & terapeuter i dit område
Terapi og behandling
Der er ingen årsagsbehandlinger tilgængelige for patienter med Holt-Oram syndrom. Der er håb om en årsagssammenhæng i fremtiden, da genterapi i øjeblikket er genstand for medicinsk forskning. Så længe denne type terapi ikke når den kliniske fase, er Holt-Oram syndrom imidlertid en uhelbredelig sygdom.
I øjeblikket er der kun symptomatiske terapimuligheder til behandling af patienter. Terapien er baseret på symptomerne i det enkelte tilfælde. Den tidlige korrektion af hjertedefekten er af særlig relevans. Denne korrektion foretages kirurgisk. I tilfælde af en Vohof-septaldefekt sigter den kirurgiske procedure at for eksempel lukke den pågældende defekt. Det samme gælder en ventrikulær septumfejl.
Korrektioner af misdannelser på ekstremiteterne er oprindeligt af sekundær betydning. Efter vellykket korrigering af hjertedefekten kan rekonstruktive kirurgiske procedurer gendanne manglende eger og separate syndaktylier. En eksisterende skoliose bekæmpes normalt under fysioterapeutisk pleje. I særligt alvorlige tilfælde kan kirurgi til implantering af en titaniumribprotese være nødvendig.
I de fleste tilfælde er der ikke behov for nogen intervention for pectus carinatum. Af psykologiske årsager kan thorax dog omformes kirurgisk, f.eks. Efter Nuss-proceduren.
Outlook og prognose
Prognosen for Holt-Oram syndrom er gunstig. Selvom der er en genetisk defekt, kan den behandles tilstrækkeligt med aktuelle medicinske muligheder. Forventet levetid for en person med syndromet er i de fleste tilfælde ikke under gennemsnittet. I tilfælde af en alvorlig misdannelse kan der være betydelige tab i forventet levealder. Prognosen er klart forværret hos disse patienter. Hjertets aktivitet er begrænset og kan føre til et tidligt liv.
Imidlertid kan størstedelen af patienterne behandles godt og med succes. Selvom der ikke er nogen kur på grund af den tilstedeværende genetiske defekt, er der gode udsigter til forskellige korrektionsmuligheder. Hjertets aktivitet er reguleret og om muligt fuldstændigt korrigeret under en kirurgisk procedure. Selvom der kan være en permanent forringelse af livsformen i sammenligning med raske mennesker, opnås en god livskvalitet på grund af behandlingen.
Fysiske abnormiteter eller misdannelser ændres i et yderligere trin. Når barnets vækstfase er afsluttet, indledes normalt en nødvendig eller ønsket korrektion af de eksisterende misdannelser. Hvis forstyrrelserne i udviklingsprocessen fører til betydelig svækkelse, træffes der korrigerende forholdsregler i barndommen for at lindre symptomerne. På grund af de optiske ændringer kan patienten opleve psykologiske konsekvenser. Dette forværrer den samlede prognose.
forebyggelse
Indtil videre kan Holt-Oram-syndromet ikke forhindres, fordi de eksterne påvirkningsfaktorer ikke er blevet afklaret endeligt.
Efterbehandling
Da Holt-Oram syndrom er en medfødt sygdom, kan det ikke helbredes fuldstændigt. Derfor er foranstaltningerne eller mulighederne for opfølgning meget begrænsede, så den berørte først og fremmest er afhængig af tidlig opdagelse og efterfølgende behandling. Hvis patienten eller forældrene ønsker at få børn, gives genetisk rådgivning for at forhindre, at dette syndrom opstår igen.
Behandlingen af Holt-Oram syndrom er primært rettet mod behandling af hjertedefekten. Dette korrigeres ved en kirurgisk procedure, hvorved patienten skal komme sig og hvile efter proceduren. Anstrengelse eller fysisk aktivitet skal undgås. Det er ikke ualmindeligt, at fysioterapeutisk behandling kræves, og mange af øvelserne kan også udføres i dit eget hjem.
De berørte er undertiden afhængige af hjælp og støtte fra deres egen familie og venner. Dette kan også forhindre psykologiske forstyrrelser eller depression. Derudover har en sund livsstil med en sund diæt en meget positiv effekt på forløbet af Holt-Oram syndrom.
Du kan gøre det selv
Holt-Oram-syndromet kan ikke forhindres og kan heller ikke behandles med hjælp til selvhjælp. Med dette syndrom er de berørte altid afhængige af en operation til behandling af hjertedefekten for at forlænge patientens forventede levetid. Jo tidligere syndromet genkendes, jo større er chancerne for et positivt forløb af sygdommen. De andre misdannelser på kroppen skal også korrigeres kirurgisk.
Da mange af de berørte også lider af psykologiske klager eller af mindreværdskomplekser forbundet med dette syndrom, er de afhængige af psykologisk behandling. Imidlertid kan samtaler med andre patienter, dine egne forældre eller venner også styrke patientens selvtillid og således lindre de psykologiske klager. Desuden er nogle patienter afhængige af hjælp fra deres medmennesker i deres hverdag, hvor varm pleje har en meget positiv effekt på forløbet af Holt-Oram syndrom.
Da sygdommen også kan påvirke de indre organer, er patienterne afhængige af regelmæssige undersøgelser og kontroller fra forskellige læger. Dette kan for eksempel forhindre nyreproblemer. Berørte børn skal informeres om konsekvenserne og komplikationerne af sygdommen.
.jpg)

.jpg)









.jpg)

.jpg)