Ved a-N-acetylgalactosaminidase-mangel det er en lysosomal opbevaringssygdom, der forekommer meget sjældent. Det er opdelt i en ung og en voksen form.
Hvad er a-N-Acetylgalactosaminidase-mangel?

A-N-acetylgalactosaminidase-manglen eller Alpha-N-acetylgalactosaminidase-mangel kaldes også i medicin Schindlers sygdom, Schindlers sygdom eller Kanzakis sygdom udpeget. Hvad der menes er en ekstremt sjælden lysosomal opbevaringssygdom, hvis arv er autosomal recessiv. Læger skelner mellem en ung form, der forekommer hos børn og unge, og en voksen form, som voksne lider under.
Den unge form for alpha-N-acetylgalactosaminidase-mangel kaldes Schindlers sygdom, mens den voksne form kaldes Kanzakis sygdom. Men der er også underinddelinger i Schindlers sygdom type I for den ungdommelige form og Schindlers sygdom type II for den voksne form. En blanding af begge former, kendt som Schindlers sygdom type III, er også mulig.
Alle tre former for sygdommen er oligosaccharidoser (glycoproteinoser), der inkluderer fucosidose og sialinsyrelagringssygdom. Navnet Schindlers sygdom eller Schindlers sygdom for alpha-N-acetylgalactosaminidase-mangel går tilbage til den tyske genetiker Detlev Schindler.
Han var den første, der beskrev sygdommen i 1988. Et år senere beskrev den japanske læge T. Kanzaki voksenformen. Han opdagede i 1991, at en alpha-N-acetylgalactosaminidase-mangel var årsagen til sygdommen.
årsager
A-N-acetylgalactosaminidase-manglen er forårsaget af en reduceret aktivitet af enzymet alfa-N-acetylgalactosaminidase. Missense- eller nonsensmutationer på NAGA-genet, der koder for alfa-N-acetylgalactosaminidase, er ansvarlige for enzymdefekten.
NAGA-genet er lokaliseret på kromosom 22-gen locus q11. Enzymet alpha-N-acetylgalactosaminidase har til opgave at katalysere spaltningen af N-acetylgalactosamin fra forskellige glycolipider og glycoproteiner. Den genetiske defekt forårsager en reduceret aktivitet af enzymet, så stoffer, der ikke metaboliseres, ophobes i kroppens celler.
I de fleste tilfælde er dette proteoglycaner, glycosphingolipider og N- eller O-bundne glycoproteiner. Akkumulering af disse forårsager skade på cellen og de berørte organer. Alpha-N-acetylgalactosaminidase-mangel er så sjælden, at der ikke er nogen nøjagtig information om dens hyppighed.
Indtil videre vides det kun tolv patienter fra otte familier, der har udviklet Schindlers sygdom i verden. Neurologiske klager forekommer ikke hos hver patient. Nogle læger har mistanke om påvirkningen af yderligere faktorer for forekomsten af neurologiske symptomer. Der er dog stadig ingen beviser, der støtter denne antagelse.
Du kan finde din medicin her
➔ Medicin mod smerterSymptomer, lidelser og tegn
Symptomerne på alfa-N-acetylgalactosaminidase-mangel afhænger af den særlige form for sygdommen. I forbindelse med Schindlers sygdom type I forekommer progressiv muskelhypotoni i det første leveår. Derudover lider de ramte babyer af en udtalt psykomotorisk regression.
Forstyrrelser i sekvensen af bevægelser, spastisk tetraplegi og myoklonale cerebrale anfald blev også registreret som symptomer. Der er også en risiko for, at barnet bliver blind. Hvis der er en voksen form for alfa-N-acetylgalactosaminidase-mangel, dvs. Schindlers sygdom type 2, forekommer symptomer, der ligner Fabrys sygdom.
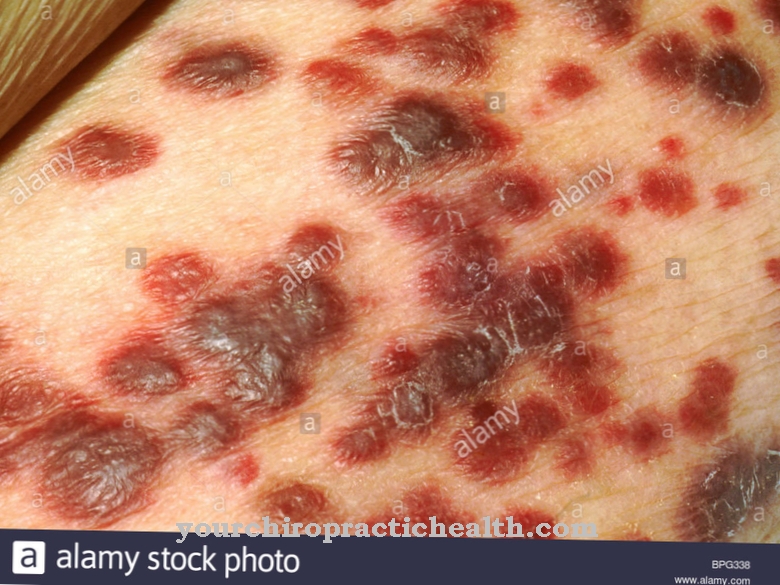
Patienter har angiokeratomer, som er godartede hudændringer. Mange patienter er let psykisk handicappede. En klinisk mellemform er Schindlers sygdom type III De ramte lider af neurologiske funktionsfejl, cerebrale anfald og psykiske handicap.
Mindre svære former er imidlertid også mulige, som er forbundet med milde psykiatriske og neurologiske klager, forsinket sprogudvikling eller symptomer, der ligner autisme.
Diagnose & kursus
Diagnosen af en alpha-N-acetylgalactosaminidase-mangel er mulig gennem påvisning af en reduceret NAGA-aktivitet. Til dette formål udføres enzymtest i blodplasma, leukocytter eller dyrkede fibroblaster eller lymfoblaster. Den kromatografiske detektion af oligosaccharider i urinen muliggør også diagnosen enzymmangel.
En DNA-analyse kan også udføres, men dette er ikke nødvendigt i de fleste tilfælde. Under graviditet kan prenatal diagnose af enzymmangel også udføres ved hjælp af en mutationsanalyse af NAGA-genet efter en chorionisk villusprøvetagning eller fostervandsprøver. Den respektive form for sygdommen kan imidlertid ikke bestemmes.
Den differentielle diagnose af Fabry-syndrom, panthothenatkinase-associeret neurodegeneration og infantil neuroaxonal dystrofi, som også har enzymdefekter, er også vigtig. Forløbet af alpha-N-acetylgalactosaminidase-mangel afhænger af den respektive sygdomsform.
Schindlers sygdomstype I betragtes som ugunstig, medens prognosen for de to andre typer er mere gunstig. Antallet af syge mennesker er imidlertid så lille, at der ikke kan gives nogen meningsfulde udsagn om forventet levealder.
Komplikationer
Symptomerne og komplikationerne af A-N-acetylgalactosaminidase-mangel afhænger meget af dens form.I de fleste tilfælde forekommer der dog bevægelsesforstyrrelser, så de berørte er relativt begrænset i deres hverdag. Kramper forekommer også, som er forbundet med svær smerte.
Babyer lider især af alvorlige udviklingsforstyrrelser, som kan føre til spastisitet hos barnet. I nogle tilfælde fører A-N-acetylgalactosaminidase-manglen til fuldstændig blindhed hos patienten. Regression af intelligens fører til retardering og dermed til handicap.
Disse begrænser normalt patientens hverdag betydeligt og har en negativ indvirkning på livskvaliteten. Ofte er de berørte afhængige af hjælp fra andre mennesker. Retardering kan også føre til en ordfindingsforstyrrelse eller en sprogforstyrrelse. Det er ikke muligt at behandle A-N-acetylgalactosaminidase-manglen årsagsmæssigt.
Af denne grund skal den berørte person tage antibiotika. Visse komplikationer skal også undgås direkte. For eksempel er lungebetændelse en af disse. Desuden kan patienten også være afhængig af kunstig ernæring.
Hvornår skal du gå til lægen?
A-N-acetylgalactosaminidase-mangel skal bestemt behandles af en læge. Denne mangel forsvinder ikke af sig selv, og der er ingen spontan heling af denne sygdom. Under alle omstændigheder skal en læge konsulteres, hvis forældrene bemærker et fald i motoriske og mentale evner hos deres barn eller baby. Klager over enkle bevægelser eller processer kan også indikere A-N-acetylgalactosaminidase-manglen og bør undersøges af en læge.
Desuden er forskellige spastisiteter også et tegn på en sygdom. Det er ikke ualmindeligt, at patienter lider af intellektuel handicap og andre funktionsfejl. Hvis disse klager er til stede, er behandling bestemt nødvendig. Hvis der ikke er nogen behandling, kan dette føre til betydelige klager og komplikationer i voksen alder og dermed reducere livskvaliteten for den berørte person betydeligt.
Svært forsinket taleudvikling kan også være et tegn på A-N-acetylgalactosaminidase-mangel og skal derfor undersøges. Som regel kan en læge konsulteres for at bestemme A-N-acetylgalactosaminidase-manglen. Den videre behandling af de enkelte klager udføres derefter af en specialist.
Læger & terapeuter i dit område
Behandling og terapi
En kausal terapi for alfa-N-acetylgalactosaminidase-mangel er ikke mulig. Derfor er den medicinske tilgang begrænset til behandling af symptomerne. Dette inkluderer en fornuftig diæt og væskeindtagelse, forebyggelse af infektionssygdomme, som også kan gøres ved indgivelse af antibiotika, samt at begrænse cerebrale angreb ved indgivelse af antiepileptika.
Yderligere behandlingstrin inkluderer fysioterapimæssige foranstaltninger til at forhindre lungebetændelse og kontrakturer og for at reducere smerter eller spastisitet ved hjælp af medicin. Desuden kan aspirationsprofylakse finde sted gennem kunstig fodring.
Nylige undersøgelser har vist, at a-N-acetylgalactosaminidase-manglen er en proteinfoldningsforstyrrelse, så enzymerstatningsterapi diskuteres som en fornuftig behandlingsforanstaltning. Genterapi er en anden tænkelig terapeutisk fremgangsmåde til behandling af lysosomale oplagringsforstyrrelser. Da Schindlers sygdom arves på en autosomal recessiv måde, anbefales genetisk rådgivning af de berørte familier.
Outlook og prognose
A-N-acetylgalactosaminidase-manglen kan føre til forskellige symptomer. I de fleste tilfælde fører manglen imidlertid til forsinket psykomotorisk udvikling. I voksen alder er patienten ofte afhængig af hjælp fra andre mennesker og kan ikke klare hverdagen alene. Bevægelsesforstyrrelser forekommer også. Dette kan bemærkes ved en halte eller af forstyrrelser i koordineringen.
I værste tilfælde kan patienten blinde eller lide af alvorlige synsforstyrrelser på grund af A-N-acetylgalactosaminidase-manglen. Det er ikke ualmindeligt, at intellektuelle handicap forekommer, der i høj grad reducerer livskvaliteten for den pågældende. Taleforstyrrelser og ordfindingsforstyrrelser forekommer også, hvilket kan påvirke livet. Børn er især påvirket af mobning og drilleri på grund af symptomerne på A-N-acetylgalactosaminidase-mangel.
En kausal behandling af A-N-acetylgalactosaminidase-manglen er ikke mulig, så behandlingen primært tager sigte på at reducere symptomerne. Forskellige terapeutiske foranstaltninger kan bruges til at reducere udviklingsforstyrrelser. I mange tilfælde er patienten dog afhængig af hjælp udefra i hverdagen. Levealderen påvirkes ikke af manglen.
Du kan finde din medicin her
➔ Medicin mod smerterforebyggelse
Der er ingen kendte forebyggende foranstaltninger mod alfa-N-acetylgalactosaminidase-mangel. Schindlers sygdom er en af de ekstremt sjældne sygdomme.
Efterbehandling
Da A-N-acetylgalactosaminidase-mangel er en medfødt sygdom, der ikke kan behandles kausalt, men kun symptomatisk, er en fuldstændig behandling ikke mulig. Den berørte person er afhængig af livslang behandling, hvorfor mulighederne for efterpleje er meget begrænsede.
Som regel er patienten ofte afhængig af indtagelse af medicin og antibiotika på grund af A-N-acetylgalactosaminidase-manglen. Der skal tages hensyn til mulige interaktioner med anden medicin, selvom antibiotika ikke bør tages sammen med alkohol. Hvis du har et angreb, skal du gå til hospitalet eller ringe til en akutlæge direkte.
Desuden skal lungerne skånes for at undgå betændelse. Derfor bør patienter med A-N-acetylgalactosaminidase-mangel under ingen omstændigheder ryge. En sund kost og en generelt sund livsstil har en meget positiv effekt på sygdomsforløbet.
Hvis patienten ønsker at få børn, er genetisk rådgivning nyttig for at forhindre, at sygdommen overføres til børnene. Da patienter ofte lider af begrænset mobilitet, er fysioterapi nyttigt for at lindre dette. Mange øvelser kan også udføres i dit eget hjem.
Du kan gøre det selv
En eksisterende A-N-acetylgalactosaminidase-mangel er en uhelbredelig sygdom. Medicinsk behandling er derfor vigtig. Selvbehandlingsforanstaltninger kan kun baseres på symptomerne for at gøre hverdagen lettere. Det er vigtigt, at forældrene observerer barnet nøje.
Forældre kan bedst hjælpe deres barn gennem forståelse og tålmodighed, kærlighed og omsorg. Yderligere er ergoterapi- og taleterapibehandlinger altid baseret på et dobbelt koncept: terapi på stedet med specialist og udførelse af øvelserne derhjemme. Forældre skal være aktive her konsekvent og med tålmodighed. Der skal sikres en diæt rig på vitaminer og mineraler, regelmæssig motion og masser af frisk luft. Dette styrker immunforsvaret og reducerer risikoen for infektion. Hvis personen har en tendens til at beslaglægge, skal de ikke være alene i lang tid, og de skal kontrolleres for at se, om de kan skade sig selv, hvis et anfald opstår.
Det meste af tiden kan patienter ikke klare hverdagen selv og kræve konstant pleje. Hvis forældre ikke har råd til dette - især med ældre børn og voksne - skal de ikke være bange for at søge hjælp. Dette kan have form af en sygeplejerske eller placering i en passende facilitet. Hvis du stadig ønsker at få børn, anbefales en konsultation med en specialist, da sygdommen er arvet.


.jpg)
.jpg)














.jpg)