Det Enzymerstatningsterapi anvendes til behandling af lysosomale opbevaringssygdomme, hvor manglen på enzymer fører til en patologisk ophobning af nedbrydningsprodukter i lysosomerne i cellerne.
De manglende enzymer på grund af genetiske defekter kompenseres for med regelmæssige intravenøse infusioner. Fordi de infunderede syntetiske enzymer ikke kan krydse blod-hjerne-barrieren på grund af deres molekylære størrelse, fungerer behandlingen kun for lysosomale opbevaringssygdomme, der ikke påvirker centralnervesystemet.
Hvad er enzymerstatningsterapi?

Lysosomer er specielle celleorganeller, hvor fremmede og endogene stoffer nedbrydes og delvist genanvendes. Specifikke hydrolyserende enzymer er nødvendige for nedbrydning og transport af stofferne. Dette er proteaser, nukleaser, lipaser og transporteringsstoffer.
Et antal kendte genetiske defekter kan føre til en svigt af visse enzymer, således at nogle nedbrydningsprodukter akkumuleres i lysosomerne i patologiske mængder og akkumuleres, indtil de når den ekstracellulære matrix, dvs. de intercellulære rum, på en ukontrolleret måde. Alle genetiske defekter, der fører til fiasko af mindst en nødvendig hydrolase, opsummeres under betegnelsen lysosomal opbevaringssygdom. Enzymerstatningsterapi (ERT, enzymerstatningsterapi) bruges til at erstatte de manglende endogene enzymer med syntetisk producerede enzymer.
Da hydrolaser består af relativt store molekyler, kan de ikke absorberes fra tarmen uden først at blive nedbrudt og inaktiveret, så de kun kan administreres via intravenøs infusion. Imidlertid forhindrer størrelsen af enzymmolekylerne også blod-hjerne-barrieren i at krydses, så behandlingen kun kan være effektiv i lysosomale opbevaringssygdomme, der ikke påvirker centralnervesystemet (CNS).
Funktion, effekt & mål
Over 50 forskellige lysosomale metaboliske lidelser er kendt, som hver kan spores tilbage til en monogenetisk defekt. De lysosomale opbevaringssygdomme kan opdeles i syv forskellige klasser afhængigt af de overdrevent lagrede stoffer på grund af den eksisterende enzymdefekt.
Mucopolysaccharidoser og oligosaccharidoser er primært egnede til en ERT. Målet med ERT er altid at kompensere for den specifikke enzymmangel ved hjælp af de kunstigt tilførte enzymer for at bringe sygdommen til stilstand eller i det mindste et mildere forløb. I detaljer er erstatningsenzymer tilgængelige for de følgende lysosomale opbevaringssygdomme:
- Gauchers sygdom
- Pompesygdom
- Fabry sygdom
- Hurler-Pfaundler syndrom (mucopolysaccharidosis I)
- Hunter's sygdom (mucopolysaccharidosis II)
• Maroteaux-Lamy syndrom (mucopolysaccharidose VI) • Niemann-Pick B
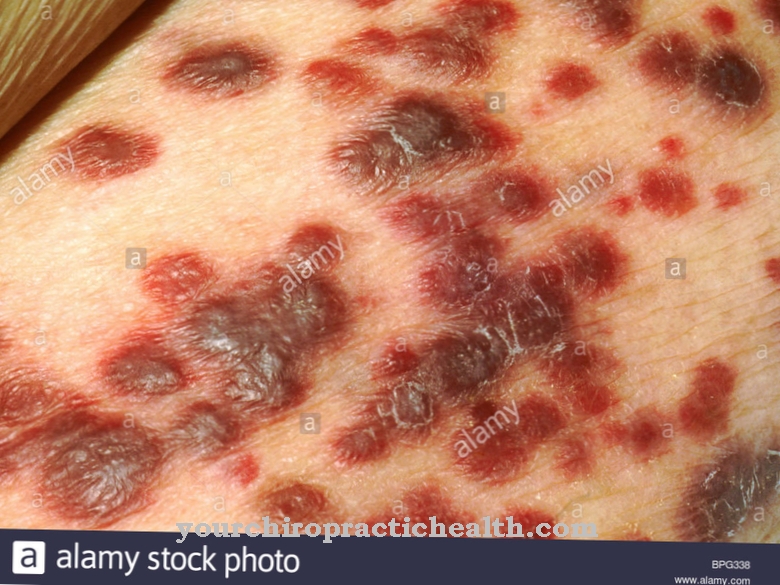
Gauchers sygdom er den mest almindelige lysosomale lagersygdom. Det forekommer i tre forskellige varianter, hvoraf to også påvirker nervesystemet. I den ikke-neuropatiske form påvirkes milten især, hvilket i høj grad forstørres og fører til sekundær skade, såsom anæmi og skade på knoglemarven. Typiske symptomer er knogle- og leddsmerter og kredsløbssygdomme. Den akutte neuropatiske variant af sygdommen viser et alvorligt forløb og giver ringe chance for at overleve ud over de to første leveår.
Lagringssygdommen Pompes sygdom skyldes en mangel på enzymet alpha-1,4-glucosidase, som er involveret i et stort antal metaboliske processer. Pompesygdom fører til en enorm forstørrelse af hjertet (hjertesygdom) og hjertesvigt. Der er tidlige, seriøse kurser, der vises i de første måneder af livet, såvel som mildere former, der først vises i senere leveår.
Fabry sygdom er forårsaget af en X-bundet genetisk defekt, så kun drenge og mænd kan blive påvirket af opbevaringssygdommen. Sygdommen fører normalt til symptomer i avanceret barndom, herunder smerteranfald, keratomer i huden, nyreproblemer og hjertemuskelskade. Manglen på enzymet alfa-galactosidase A fører til en ophobning af ceramidtrihexosid, hvilket er årsagen til udløsningen af symptomer, som også kan påvirke det autonome nervesystem.
Det er ikke ualmindeligt, at skaden kan føre til et hjerteanfald, nyreinfarkt eller endda et slagtilfælde. Hurler-Pfaundler syndrom er også kendt som mucopolysaccharidose, type I og er forårsaget af en forstyrrelse af glycosaminoglycan metabolismen. Sygdommen er forbundet med en lang række symptomer, herunder alvorlig psykisk svækkelse og alvorlige skeletændringer. Sygdomsforløbet er alvorligt, så den gennemsnitlige forventede levealder gives som 11 til 14 år. Hunter's sygdom svarer til mucopolysaccharidosis, type 2 og er - ligesom Hurlers sygdom - forårsaget af en X-bundet defekt. Sygdommen er kendetegnet ved kurser med varierende sværhedsgrad, fra at forekomme i den tidlige barndom til milde kurser, der kun forekommer hos voksne mænd.
På grund af de mest almindelige hjertesymptomer, såsom hjerteklaffedefekter og hjertemuskelproblemer, varierer levealderen fra normal til lidt begrænset. Maroteaux-Lamy syndrom (MPS VI) er en af mucopolysaccharidoserne, der arves som en autosomal recessiv egenskab, fordi den genetiske defekt, der forårsager det, ikke er på X-kromosomet. Sygdommen er meget sjælden med et tilfælde pr. 455.000 fødsler. Der er kendte milde og svære former.
Symptomerne er forstørret lever og milt, karpaltunnelsyndrom og ændringer i hjerteklapperne. Niemann-Pick B er en sfingomyelin lipidose, som er en af de lysosomale opbevaringssygdomme og er forårsaget af en genetisk defekt på kromosom 11. Mens type B af sygdommen hovedsageligt påvirker leveren og milten, har type A også betydelige neuronale problemer.
Du kan finde din medicin her
➔ Medicin mod smerterRisici, bivirkninger og farer
Da mange af de lysosomale opbevaringssygdomme, der kan behandles med enzymerstatningsterapi, tager et alvorligt forløb med en tilsvarende højere dødelighed, hvis de ikke behandles, er den største risiko i ERT, at det valgte erstatningsenzym ikke fungerer eller fungerer for svagt.
En anden risiko ligger mindre i selve behandlingen end i det faktum, at den underliggende sygdom genkendes for sent, så ERT kan stoppe i løbet af kurset, men skaden, der allerede er forårsaget, kan ikke regressere. Cirka hver anden patient, der behandles midlertidigt, reagerer på infusionerne med symptomer som feber og kulderystelser. Årsagerne hertil er endnu ikke fuldt ud forstået. Nogle patienter reagerer ved at danne antistoffer, og der har været kendte tilfælde, hvor patienter har reageret med udslæt og bronchospasme.















.jpg)