Det Aicardi-Goutières syndrom er en ekstremt sjælden, autosomal recessiv nedarvet genmutation, der udløser en genetisk heterogen udviklingsforstyrrelse i hjernen hos omkring tre måneder gamle spædbørn og ud over alvorlige motoriske svækkelser er forbundet med spasticitet og epilepsi. For svære fænotyper er en maksimal levealder på ti år en retningslinje, selvom mildere fænotyper med RNASEH2B-mutationer muligvis kan leve til at være mere end ti år.
Hvad er Aicardi-Goutières-syndrom?

Lægen forstår, at Aicardi-Goutières-syndrom er en autosomal recessiv arvelig sygdom, der svarer til en genetisk heterogen udviklingsforstyrrelse i hjernen hos babyer omkring tre måneders alder. Klinisk har syndromet et lignende udseende som en intrauterin infektion, dvs. en infektion, der blev erhvervet i livmoderen, men ingen patogener kan detekteres, da der er en genetisk årsag.
Visse cellekernenzymer er mindre aktive end sædvanligvis på grund af den genetiske defekt, og renser derfor kromosomerne markant mindre for falske RNA-proteiner. Dette fører til en ophobning af DNA-segmenter i cellerne. Da dette får cellerne til at dø, bliver immunsystemet aktivt og forårsager betændelse i de relevante områder.
I 1984 beskrev de franske læger Françoise Goutières og Jean François Aicardi først dette fænomen. Siden da er færre end 150 tilfælde blevet kendt, så den genetiske defekt er tilsvarende sjælden. De enkelte tilfælde er hidtil mest set i familier, da et andet barn også vil udvikle syndromet med en 25 procent sandsynlighed.
årsager
Der er ingen erhvervet form for Aicardi-Goutières syndrom. Hjernens udviklingsforstyrrelse er altid medfødt og kan spores tilbage til en autosomal recessiv genmutation. For at udløse et sygdomsudbrud skal det defekte gen være til stede på begge sider af arven. Imidlertid er nye mutationer også mulige. Indtil videre er fem genplaceringer blevet knyttet til mutationen.
Disse inkluderer TREX1, RNASEH2A, RNASEH2B og RNASEH2C samt SAMHD1. De første beskrivelser af syndromet antager en årsag i forbindelse med genet TREX1 på kromosom 3p21. RNASEH2-A-generne er genloki for tre forskellige proteiner, der spiller en rolle i dannelsen af det trimeriske cellekernenzym RNase H.
Dette cellekernenzym er på sin side ansvarlig for at fjerne forkert placerede RNA-molekyle-byggesten fra DNA'et. Uden RNase H forekommer tidlig embryonal død. Hvis cellekernenzymet derimod er til stede, men ikke fuldt aktiv, lagres individuelle fragmenter af visse gener i kroppens celler. De forårsager apoptose, som udløser en inflammatorisk proces baseret på den naturlige immunreaktion.
Du kan finde din medicin her
➔ Medicin mod muskelkramperSymptomer, lidelser og tegn
Børn med Aicardi-Goutières-syndrom er vanskelige at fodre. Du har uønskede bevægelser i øjnene og svirrende og lider af lettere anfald af feber og opkast. Lidt mindre end halvdelen af de berørte børn mister de motoriske færdigheder, de har lært indtil nu efter ca. seks måneder. Med hensyn til motorik forekommer især pyramidetegn.
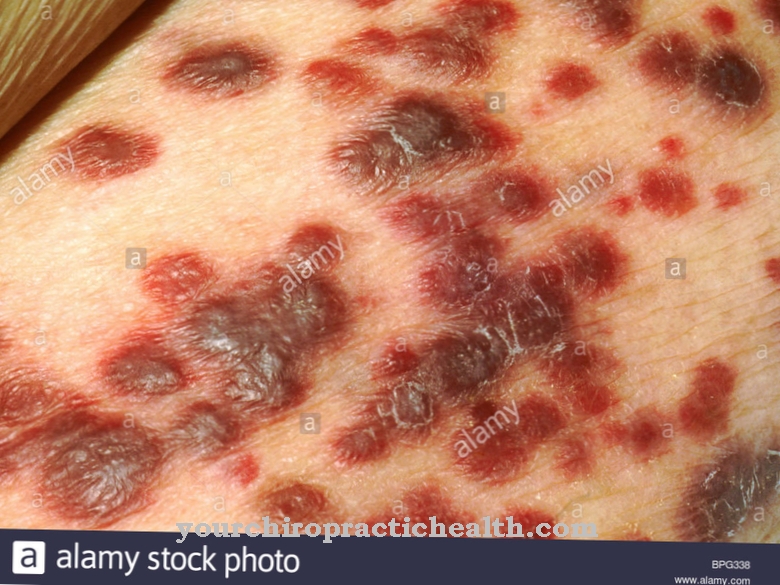
Ud over spastisk lammelse kan der være ukoordinerede bevægelser, der får musklerne i arme og ben til at trække sig sammen eller forårsage anfald. Ud over forstyrrelse af muskeltonus udvides undertiden organer som lever og milt. Senere er der en stigende forsinkelse i psykomotorisk udvikling. Lejlighedsvis forekommer også frostskadelignende hudændringer.
Diagnose & kursus
Ved den differentierede diagnose skal lægen primært udelukke Toxoplasma-parasitter, hvis der er mistanke om Aicardi-Goutières-syndrom. Computertomografi viser forkalkninger i basalganglier på begge sider af AGS. Tab af hjernestoffer og misdannelser i hjernestoffet kan også være tegn på computertomografi.
Nervevandsundersøgelsen viser et forhøjet CSF-lymfocytoseniveau og alfa-interferoner, hvilket indikerer en inflammatorisk proces i det centrale nervesystem. I serum hos AGS-patienter reduceres blodpladerne, og samtidig øges leverenzymværdierne. Hvad angår sygdomsforløbet, dør mange af de berørte stadig i barndommen, idet ti år er den maksimale forventede levealder for særlig alvorlige former for mutation.
Patienter med mildere former lever ofte at være over ti år gamle. Disse mildere fænotyper svarer for det meste til RNASEH2B-mutationer, som er til stede i næsten halvdelen af alle tilfælde. Sygdommen udvikler sig i begyndelsen over flere måneder, indtil der er flere handicap. Efter disse første måneder stabiliseres sygdommen imidlertid normalt på et jævnt forløb. Skønt næsten alle patienter over ti år måtte kæmpe med alvorlige multiple handicap, har der i individuelle tilfælde været betydeligt mildere kurser.
Komplikationer
Aicardi-Goutières syndrom har alvorlige komplikationer, der kan have en meget negativ indflydelse på mennesker og børn. I de fleste tilfælde viser syndromet sig vanskeligt med fodring og øjenbevægelser, der er jerky og ukontrolleret. Efterhånden som sygdommen skrider frem, bliver feber og opkast mere almindelige.
Med Aicardi-Goutières-syndrom viser børnene spastisk lammelse af arme og ben, så de ikke længere kan koordinere og bevæge dem korrekt. I værste fald kan der forekomme anfald, hvilket kan være særlig farligt for børn. Aicardi-Goutières syndrom fører også til mental retardering.
I de fleste tilfælde fører symptomerne til død i barndommen, så forventet levealder falder enormt. Hvis barnet ikke dør med Aicardi-Goutières syndrom, vil der være betydelige komplikationer og vanskeligheder i hverdagen. På grund af de fysiske og psykologiske begrænsninger er patienten altid afhængig af hjælp fra andre mennesker og plejere.
Et ophold på hospitalet er normalt nødvendigt. Aicardi-Goutières-syndrom kan også påvirke leveren og milten, hvilket får dem til at forstørre. Dette resulterer normalt i smerter i de respektive organer.
Hvornår skal du gå til lægen?
I de fleste tilfælde kan Aicardi-Goutières syndrom ikke behandles af en læge. Patienten dør efter de første ti leveår. Som regel bestemmes symptomerne allerede af en børnelæge. Forældre kan dog kontakte en børnelæge, hvis der er mærkbare forsinkelser i udviklingen eller i udviklingen af forskellige typer spasticitet.
Et besøg hos lægen er nødvendigt, selv med rykkende bevægelser og en stærk og vedvarende feber. Normalt kan medicin begrænse feber og opkast. Det er ikke ualmindeligt, at børnene lider af lammelse og kramper. Disse kan også begrænses ved behandling, så det i disse tilfælde også anbefales at besøge lægen.
En akutlæge kan også kaldes i akutte nødsituationer eller anfald. Det er ikke ualmindeligt, at forældre lider af psykologiske klager eller depression. I dette tilfælde anbefales den psykologiske behandling af forældrene. Dette bør gives psykologisk støtte, især efter at barnet dør af Aicardi-Goutières syndrom.
Læger & terapeuter i dit område
Behandling og terapi
Aicardi-Goutières syndrom kan endnu ikke behandles kausalt, men behandles kun symptomatisk. I denne sammenhæng kan anfald forebygges ved indgivelse af for eksempel antiepileptika. Fysioterapeutiske foranstaltninger anvendes normalt til at reducere spasticitet og kontrakturer.
Visse salver og cremer kan bruges mod hudsymptomer. Terapimulighederne inkluderer også tidlig indgriben, hvor ikke kun de berørte børn, men især forældrene, plejes.
Outlook og prognose
Aicardi-Goutières-syndrom fører til forskellige misdannelser og stress for patienten. Især barnets forældre påvirkes ofte af svære psykologiske klager og depression og har derfor brug for psykologisk støtte.
Barnet selv lider af alvorlige handicap og epileptiske anfald. Dette fører til intens feber og opkast. Ofte lider barnet også af ukontrollerbare bevægelser i øjnene og lammelse i forskellige dele af kroppen. Derudover kan der forekomme anfald, som ofte er forbundet med smerter. Barnets udvikling er stærkt begrænset af Aicardi-Goutières-syndromet, og motoriske forstyrrelser forekommer. Syndromet påvirker også det centrale nervesystem negativt. Patientens livskvalitet er ekstremt begrænset, og den pågældende er normalt afhængig af hjælp fra andre mennesker i hverdagen.
Aicardi-Goutières-syndrom kan ikke behandles, så kun symptomerne kan begrænses. De epileptiske anfald kan reduceres relativt godt ved hjælp af medicin. Forskellige behandlinger er også tilgængelige for patienten.
Du kan finde din medicin her
➔ Medicin mod muskelkramperforebyggelse
Et foster kan testes for Aicardi-Goutières syndrom, mens det stadig er i livmoderen ved at tage en fostervandsprøve. Forventer forældre kan derefter beslutte mod barnet. Ingen andre forebyggende foranstaltninger er tilgængelige, fordi syndromet er en mutation.
Efterbehandling
Opfølgningsmuligheder er relativt begrænset til Aicardi-Goutières syndrom. Patienten er altid afhængig af medicinsk behandling for at lindre symptomerne og undgå mulige komplikationer. Som regel reduceres den forventede levetid også betydeligt af syndromet.
Da tilstanden er arvelig, kan den ikke behandles fuldstændigt. En rent symptomatisk behandling er tilgængelig for patienten. Hvis du vil have børn, kan genetisk rådgivning også udføres for at forhindre gentagelse af Aicardi-Goutières syndrom. De berørte er mest afhængige af at tage medicin.
Disse skal tages regelmæssigt, og de mulige interaktioner med andre lægemidler bør også overvejes. Børnene får passende behandling og støtte for at modvirke spastisiteten. De kan dog ikke helbredes fuldstændigt.
Kontakt med andre syge af Aicardi-Goutières syndrom har ofte en positiv effekt på det videre sygdomsforløb. Det er ikke ualmindeligt, at information udveksles, og hverdagen gøres lettere. Endvidere er forældrene ofte også afhængige af psykologisk behandling. Samtaler med venner eller bekendte kan også være nyttige.
Du kan gøre det selv
Aicardi-Goutières syndrom er en sygdom med flere handicap. Forældre bliver nødt til at søge permanent medicinsk behandling med deres barn. Byrden for forældrene er meget høj, og psykologisk støtte anbefales stærkt. Yderligere professionel hjælp til den daglige pleje af barnet bør også søges uden tøven.
Hvis der er andre børn i familien, bør muligheden for at anbringe dem i en pleje også diskuteres. Da det syge barn har brug for intensiv pleje, bør byrden ikke undervurderes. For forældre med et sygt barn er risikoen for depression især højere. At tage sig af ens eget helbred - fysisk og mentalt - bør ikke overses. Forældre kan kun være en stabil støtte til deres barn, hvis de selv er stabile.
Krampeanfald og hyppig opkast kræver konstant observation af barnet. Barnet har brug for pleje af forældre eller professionel til alle aktiviteter i det daglige liv.
Forældre skal sikre tilstrækkelig bevægelse gennem motoriske øvelser, gøre barnets diæt rig på vitale stoffer og let fordøjelig og sikre tilstrækkeligt væskeindtag. Fysioterapeutisk støtte er især vigtig for at reducere spastiske anfald og ledsager lægemiddelterapi med antiepileptika. Forældre kan behandle dermatoser støttende ved omhyggeligt at påføre cremer og salver.

.jpg)
.jpg)













.jpg)