Neurologi er et af de mest mangefacetterede og fascinerende medicinområder Ud over sygdomme som multippel sklerose, Alzheimers sygdom og det velkendte slagtilfælde er der også sådanne Spinocerebellare ataksier af enorm betydning. Disse repræsenterer den generiske betegnelse for en lang række forstyrrelser i koordination af bevægelser Tabet af nerveceller fører til en defekt interaktion mellem musklerne.
Hvad er spinocerebellær ataksi?
© designua - stock.adobe.com
Spinocerebellare ataksier (Engelsk: spinocerebellare ataksier, kort: SCA) betegner en gruppe neurodegenerative sygdomme i centralnervesystemet (CNS) hos mennesker. Neuronerne (nerveceller) i lillehjernen (cerebellum) og rygmarven (medulla spinalis) omkommer gradvist. Sygdomme af denne type er ekstremt sjældne, de forekommer i USA og Centraleuropa med en gennemsnitlig hyppighed på en ny sygdom pr. Hundrede tusinde indbyggere.
årsager
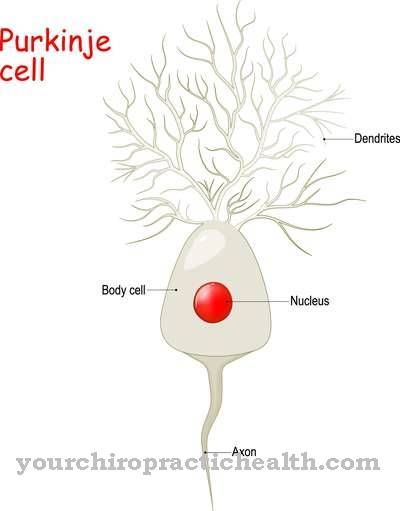
Årsagen ligger i døden af Purkinje-cellerne (de største neuroner i lillehjernen) på grund af den autosomale dominerende arv fra patologiske gener. Mere end 25 forskellige genplaceringer er i øjeblikket kendt. Undergrupper af spinocerebellær ataksi er defineret i henhold til disse udløsende gener og benævnt SCA type 1, type 2, type 3 eller SCA1, SCA2, SCA3 et cetera.
Typerne 1, 2, 6, 7 og 17 hører til gruppen af trinucleotidsygdomme (såsom Huntingtons sygdom), da sygdommen er forårsaget af en mutation i form af en usædvanlig lang triplet-gentagelse (triplet = tre på hinanden følgende nukleobaser af en nukleinsyre) i kodonen CAG (hvad svarer til aminosyren glutamin). Spinocerebellær ataksi type 3 (SCA3), også kendt som Machado-Joseph sygdom (MJD), er den mest almindelige form for denne sygdom i Tyskland og tegner sig for 35 procent af de autosomale dominerende arvelige cerebellare ataksier.
Symptomer, lidelser og tegn
I de fleste tilfælde er sygdommens begyndelse mellem 30 og 40 år. Kardinalsymptomet er koordinationsforstyrrelsen i bevægelsessekvenser (ataksi). Patienter klager over den resulterende ustabilitet, når de går og står, såvel som klodset taget genstande.
Derudover er der en ændring i melodien til tale (dysarthria) og en bevægelsesforstyrrelse i øjnene (nystagmus). Afhængigt af undergrupper af spinocerebellær ataksi er der også symptomer, der forekommer afhængigt af inddragelse af andre hjerneområder, for eksempel muskelkramper, spastisitet (patologisk stigning i muskeltonus).
Også hukommelsesforstyrrelser (demens), sanseforstyrrelser og unormale fornemmelser, slukningsforstyrrelser, inkontinens, forringelse af synet, aftagelse af bevægelsessekvenser og rastløse ben (rastløse ben syndrom) Nogle patienter har Parkinsons-lignende symptomer, der reagerer godt på lægemidler, der bruges til behandling af Parkinsons sygdom.
Diagnose & sygdomsforløb
Diagnosen stilles på grundlag af den detaljerede anamnese, klinisk-neurologiske undersøgelser og yderligere fund (f.eks. CSF-undersøgelse, magnetisk resonansafbildning og neurofysiologisk undersøgelse) for at udelukke andre mulige sygdomme. En molekylær genetisk undersøgelse er presserende nødvendigt for at bekræfte diagnosen.
Det er ofte vanskeligt, undertiden umuligt, at bestemme, hvilken type ataksi der er, fordi de kun er lidt forskellige. Efterhånden som sygdommen skrider frem, øges symptomerne, indtil sygdommen fører til død (i de fleste tilfælde).
Komplikationer
Spinocerebellare ataksier kan forårsage forskellige komplikationer, afhængigt af sygdommens form. Generelt fører ataksier til muskelspasmer, spastiske rykker og ændringer i melodien til tale. Derudover kan hukommelsesforstyrrelser forekomme, som kan udvikle sig til demens, efterhånden som sygdommen skrider frem.
Opbremsningen af bevægelsessekvenser repræsenterer ofte en betydelig begrænsning i hverdagen for de berørte.I forbindelse med andre komplikationer, såsom den typiske forringelse af synet, forårsager ataksi undertiden også følelsesmæssig nød. Uanset sygdommens form forøges symptomerne, efterhånden som sygdommen skrider frem. I de fleste tilfælde resulterer spinocerebellare ataksier i patientens død. Behandling af en neurologisk lidelse medfører også risici.
Lægemiddelterapi er altid forbundet med visse bivirkninger og interaktioner for dem, der er berørt. Det samme gælder ergoterapi og fysioterapi, som lejlighedsvis er forbundet med spændinger, ømme muskler og mindre kvæstelser. Kirurgiske indgreb er sjældne ved spin-oprørske ataksier, men kan føre til infektioner, blødning, sekundær blødning, infektioner og nedsat sårheling. Hvis proceduren går dårligt, kan den oprindelige lidelse forværres.
Hvornår skal du gå til lægen?
Som regel med denne sygdom er patienten altid afhængig af medicinsk behandling fra en læge. Frem for alt har en tidlig diagnose med tidlig behandling en meget positiv effekt på det videre kursus. Dette er den eneste måde at forhindre yderligere komplikationer, da denne sygdom ikke kan helbrede sig selv.
En læge skal konsulteres, hvis den berørte person oplever problemer med bevægelse og koordination. Som regel kan patienter ikke let gå lige eller nå ordentligt efter tingene. Kramper i musklerne eller spastisitet kan også indikere denne sygdom. Mange af de berørte lider også af svære sværhedsgrader eller endda fra inkontinens og andre unormale fornemmelser.
Sygdommen kan påvises af en læge. For yderligere behandling er det normalt nødvendigt at besøge en specialist. Det kan heller ikke universelt forudsiges, om en komplet kur kan forekomme.
Behandling og terapi
En kausal terapi for spinocerebellare ataksier er endnu ikke kendt. Fokus er på symptomatisk behandling i betydningen funktionel vedligeholdelse for at bevare patientens livskvalitet så længe som muligt. Disse inkluderer medicin, ergoterapi og fysioterapi samt taleterapi.
Ifølge det tyske samfund for neurologi (DGN) viste en pilotundersøgelse, at cerebellare ataksier reagerer på den aktive ingrediens riluzol. Selvom forskning på spinocerebellære ataksier er blevet intensiveret i de sidste par år, er den i øjeblikket ikke så avanceret, at en kurativ terapi kan forventes i den nærmeste fremtid.
I ergoterapi og fysioterapi bevares mobiliteten i de enkelte kropssnit, svindlende muskler styrkes, og dannelsen af synapser stimuleres. Aktiviteterne i det daglige liv udøves for at bevare patientens uafhængighed så meget som muligt. Taleterapi arbejder med eksisterende sprogproblemer.
forebyggelse
Da det er en genetisk sygdom, er enhver form for forebyggelse umulig.
Efterbehandling
Samlebetegnelsen 'spinocerebellær ataksi' beskriver genetisk bestemte kliniske billeder, hvor nervesystemet påvirkes. Forstyrrede motoriske processer til demens i det sene stadium er typiske symptomer. I modsætning til andre arvelige sygdomme forekommer symptomerne ikke kun i barndommen. Ataksien bryder gennemsnitligt ud mellem 30 og 40 år, og hos nogle patienter tidligere eller ikke før de er 50 til 60 år gamle. Indtil dette tidspunkt var patienten symptomfri.
Som det står nu, kan spinocerebellare ataksi ikke heles. Sygdommen er kronisk og har under alle omstændigheder et dødeligt resultat. Opfølgningspleje er at lindre symptomer og sætte patienten i stand til at leve et stort set normalt liv. En parallel, psykoterapeutisk akkompagnement er nyttig for den pågældende, da sygdommen kan være forbundet med følelsesmæssig stress.
Pårørende har også muligheden for at modtage støtte fra en psykoterapeut. Øvelserne skal bevare lemmernes mobilitet. Hvis sprogcentret påvirkes af de neurologiske svigt, anbefales taleterapibehandlinger. Opfølgningsforanstaltningerne er langvarige, de ledsager patienten fra udbruddet til det sene stadie af sygdommen. Opfølgningspleje giver kun mening, hvis det er blevet udført konsekvent i årevis.
Du kan gøre det selv
Ved spinocerebellær ataksi er fokus på medicinsk og fysioterapeutisk behandling. Derudover kan patienter gøre et par ting for at gøre hverdagen lettere med sygdommen.
Koordinationsforstyrrelsen begrænser de, der er berørt i hverdagen markant. Derfor er den vigtigste foranstaltning at kompensere for begrænsningerne og at støtte den syge så godt som muligt. I de fleste tilfælde er det nødvendigt at flytte til en lejlighed egnet til handicappede. De stigende bevægelsesbegrænsninger kræver også et gåhjælpemiddel for patienten. Berørte mennesker har brug for støtte i hverdagen, da selv enkle aktiviteter som regel ikke længere kan udføres uden hjælp udefra. Lidende bør læse faglitteratur om spinocerebellær ataksi for bedre at forstå og acceptere sygdommen.
Derudover anbefales drøftelser med andre berørte personer, for eksempel inden for rammerne af en selvhjælpsgruppe for personer med Parkinson. I de senere stadier af sygdommen er ambulant og til sidst ambulant pleje nødvendig. I de sidste stadier af sygdommen, når bevægelser og samtaler bliver stadig vanskeligere, kan omfattende terapeutisk pleje af patienten og hans pårørende også være nyttig.
.jpg)
.jpg)

.jpg)

.jpg)





.jpg)